Example Scripts¶
This is a collection of example scripts for some of Ursgal’s functionality. The simple example scripts are designed to get familiar with executing engines through Ursgal. Examles of complete workflows can be modified for different needs. Further example scripts can also be found in ~ursgalexample_scripts
Simple Example Scripts¶
Simple example search¶
-
simple_example_search.main()¶ Executes a search with OMSSA, XTandem and MS-GF+ on the BSA1.mzML input_file
- usage:
- ./simple_example_search.py
Note
Myrimatch does not work with this file. To use MSAmanda on unix platforms, please install mono (http://www.mono-project.com/download)
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
import shutil
def main():
"""
Executes a search with OMSSA, XTandem and MS-GF+ on the BSA1.mzML
input_file
usage:
./simple_example_search.py
Note:
Myrimatch does not work with this file.
To use MSAmanda on unix platforms, please install mono
(http://www.mono-project.com/download)
"""
uc = ursgal.UController(
profile="LTQ XL low res",
params={
"database": os.path.join(os.pardir, "example_data", "BSA.fasta"),
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
# 'peptide_mapper_class_version' : 'UPeptideMapper_v2',
},
)
if sys.maxsize > 2 ** 32:
xtandem = "xtandem_vengeance"
else:
xtandem = "xtandem_sledgehammer"
engine_list = [
"omssa",
xtandem,
"msgfplus_v2016_09_16",
]
mzML_file = os.path.join(
os.pardir, "example_data", "BSA_simple_example_search", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
uc.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
uc.params["http_output_folder"] = os.path.dirname(mzML_file)
uc.fetch_file(
engine="get_http_files_1_0_0",
)
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
unified_file_list = []
for engine in engine_list:
unified_search_result_file = uc.search(
input_file=mzML_file, engine=engine, force=False
)
unified_file_list.append(unified_search_result_file)
uc.visualize(
input_files=unified_file_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
main()
Simple example using combined fdr (or pep)¶
-
simple_combined_fdr_score.main()¶ Executes a search with 3 different search engines on an example file from the data from Barth et al. (The same file that is used in the XTandem version comparison example.)
- usage:
- ./simple_combined_fdr_score.py
This is a simple example script to show how results from multiple search engines can be combined using the Combined FDR Score approach of Jones et al. (2009).
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Executes a search with 3 different search engines on an example file from the
data from Barth et al. (The same file that is used in the XTandem version
comparison example.)
usage:
./simple_combined_fdr_score.py
This is a simple example script to show how results from multiple search engines
can be combined using the Combined FDR Score approach of Jones et al. (2009).
"""
engine_list = [
"omssa_2_1_9",
"xtandem_piledriver",
# 'myrimatch_2_1_138',
"msgfplus_v9979",
]
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"modifications": [],
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
],
"ftp_output_folder": os.path.join(
os.pardir, "example_data", "xtandem_version_comparison"
),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
if os.path.exists(params["ftp_output_folder"]) is False:
os.mkdir(params["ftp_output_folder"])
uc = ursgal.UController(profile="LTQ XL low res", params=params)
mzML_file = os.path.join(params["ftp_output_folder"], params["ftp_include_ext"][0])
if os.path.exists(mzML_file) is False:
uc.fetch_file(engine="get_ftp_files_1_0_0")
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
validated_files_list = []
for engine in engine_list:
unified_result_file = uc.search(
input_file=mzML_file,
engine=engine,
)
validated_file = uc.validate(
input_file=unified_result_file,
engine="percolator_2_08",
)
validated_files_list.append(validated_file)
combined_results = uc.combine_search_results(
input_files=validated_files_list,
engine="combine_FDR_0_1",
# use combine_pep_1_0_0 for combined PEP :)
)
print("\tCombined results can be found here:")
print(combined_results)
return
if __name__ == "__main__":
main()
Simple de novo search¶
-
simple_de_novo_search.main()¶ Executes a search with Novor and PepNovo on the BSA1.mzML input_file
- usage:
- ./simple_de_novo_search.py
Note
PepNovo currently only works for Linux. Novor needs to be downloaded from http://rapidnovor.com/novor/standalone/ and stored at <ursgal_path>/resources/<platform>/<architecture>/novor_1_1beta
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
import shutil
def main():
"""
Executes a search with Novor and PepNovo on the BSA1.mzML
input_file
usage:
./simple_de_novo_search.py
Note:
PepNovo currently only works for Linux.
Novor needs to be downloaded from http://rapidnovor.com/novor/standalone/ and
stored at <ursgal_path>/resources/<platform>/<architecture>/novor_1_1beta
"""
uc = ursgal.UController(
profile="LTQ XL low res",
params={
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
},
)
engine_list = [
"novor_1_05",
"pepnovo",
]
mzML_file = os.path.join(
os.pardir, "example_data", "BSA_simple_de_novo_search", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
uc.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
uc.params["http_output_folder"] = os.path.dirname(mzML_file)
uc.fetch_file(
engine="get_http_files_1_0_0",
)
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
unified_file_list = []
for engine in engine_list:
unified_search_result_file = uc.search(
input_file=mzML_file, engine=engine, force=False
)
unified_file_list.append(unified_search_result_file)
uc.visualize(
input_files=unified_file_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
main()
Simple crosslink search¶
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
def main():
"""
Simple crosslink search using Kojak and an example file from Barth et al.
2014, please note that this is only testing if Kojak works.
The sample represents no crosslink data in particular, but Kojak is used to
map possible disulfide bonds.
Parameters have not been optimized yet, please use this script as a template
to use Kojak. Please note the different approach for executing Percolator.
Note:
Please note that Kojak has to installed manually at the resources folder
under engine-folder (kojak_1_5_3).
Additinally Percolator (2.08) has to be symlinked or copied to
engine-folder 'kojak_percolator_2_08'.
Usage:
./simple_srosslink_search.py <path_2_chlamydomonas_reinhardtii_database>
Note:
The peptide -.GHYLNATAGTC[-34.00]EEMMK.- from Rubisco large subunit is
detected with a predicted disulfide bond site at this position.
This site is reported to have a disulfide bond in the Uniprot database.
The modification (C +32, C -34) was set according to 'Tsai PL, Chen SF,
Huang SY (2013) Mass spectrometry-based strategies for protein disulfide
bond identification. Rev Anal Chem 32: 257–268'
Please use the reference C. reinhardtii (TaxId 3055 ) proteome from
Unipot.
"""
params = {
"database": sys.argv[1],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
],
"ftp_output_folder": os.path.join(
os.pardir, "example_data", "simple_crosslink_search"
),
"cross_link_definition": ["C C -2 test_if_kojak_runs"],
"mono_link_definition": ["C 32", "C -34"],
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
],
"precursor_min_mass": 500,
"precursor_max_mass": 8000,
"precursor_mass_tolerance_plus": 15,
"precursor_mass_tolerance_minus": 15,
"max_accounted_observed_peaks": 0, # i.e. all
"max_num_mods": 2,
}
if os.path.exists(params["ftp_output_folder"]) is False:
os.mkdir(params["ftp_output_folder"])
uc = ursgal.UController(profile="LTQ XL low res", params=params)
mzML_file = os.path.join(params["ftp_output_folder"], params["ftp_include_ext"][0])
if os.path.exists(mzML_file) is False:
uc.fetch_file(engine="get_ftp_files_1_0_0")
td_database_name = params["database"].replace(".fasta", "_target_decoy.fasta")
if os.path.exists(td_database_name) is False:
uc.execute_misc_engine(
input_files=[params["database"]],
engine="generate_target_decoy_1_0_0",
output_file_name=td_database_name,
)
uc.params["database"] = td_database_name
engine = "kojak_1_5_3"
mgf_file = uc.convert(
input_file=mzML_file,
engine="mzml2mgf",
)
raw_result = uc.search_mgf(
input_file=mgf_file,
engine=engine,
force=False,
)
for extension in uc.unodes[engine]["META_INFO"]["all_extensions"]:
if "perc" not in extension:
continue
file_to_validate = raw_result.replace(
uc.unodes[engine]["META_INFO"]["output_extension"], extension
)
try:
uc.validate(file_to_validate, "kojak_percolator_2_08")
except:
pass
return
if __name__ == "__main__":
main()
Target decoy generation¶
-
target_decoy_generation_example.main()¶ Simple example script how to generate a target decoy database.
Note
By default a ‘shuffled peptide preserving cleavage sites’ database is generated. For this script a ‘reverse protein’ database is generated.
usage:
./target_decoy_generation_example.py
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Simple example script how to generate a target decoy database.
Note:
By default a 'shuffled peptide preserving cleavage sites' database is
generated. For this script a 'reverse protein' database is generated.
usage:
./target_decoy_generation_example.py
"""
params = {
"enzyme": "trypsin",
"decoy_generation_mode": "reverse_protein",
}
fasta_database_list = [os.path.join(os.pardir, "example_data", "BSA.fasta")]
uc = ursgal.UController(params=params)
new_target_decoy_db_name = uc.execute_misc_engine(
input_file=fasta_database_list,
engine="generate_target_decoy_1_0_0",
output_file_name="my_BSA_target_decoy.fasta",
)
print("Generated target decoy database: {0}".format(new_target_decoy_db_name))
if __name__ == "__main__":
main()
Convert .raw to .mzML¶
-
convert_raw_to_mzml.main(input_path=None)¶ Convert a .raw file to .mzML using the ThermoRawFileParser. The given argument can be either a single file or a folder containing raw files.
- Usage:
- ./convert_raw_to_mzml.py <raw_file/raw_file_folder>
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
import glob
def main(input_path=None):
"""
Convert a .raw file to .mzML using the ThermoRawFileParser.
The given argument can be either a single file or a folder
containing raw files.
Usage:
./convert_raw_to_mzml.py <raw_file/raw_file_folder>
"""
R = ursgal.UController()
# Check if single file or folder.
# Collect raw files if folder is given
input_file_list = []
if input_path.lower().endswith(".raw"):
input_file_list.append(input_path)
else:
for raw in glob.glob(os.path.join("{0}".format(input_path), "*.raw")):
input_file_list.append(raw)
# Convert raw file(s)
for raw_file in input_file_list:
mzml_file = R.convert(
input_file=raw_file,
engine="thermo_raw_file_parser_1_1_2",
)
if __name__ == "__main__":
if len(sys.argv) != 2:
print(main.__doc__)
main(input_path=sys.argv[1])
Simple mgf conversion¶
-
simple_mgf_conversion.main()¶ Simple example script to demonstrate conversion for mzML to mgf file conversion
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import shutil
def main():
"""
Simple example script to demonstrate conversion for mzML to mgf file
conversion
"""
uc = ursgal.UController(
profile="LTQ XL low res",
params={},
)
mzML_file = os.path.join(
os.pardir, "example_data", "simple_mzml_to_mgf_conversion", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
uc.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
uc.params["http_output_folder"] = os.path.dirname(mzML_file)
uc.fetch_file(engine="get_http_files_1_0_0")
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
mgf_file = uc.convert(
input_file=mzML_file, engine="mzml2mgf_1_0_0" # from OpenMS example files
)
if __name__ == "__main__":
print(__doc__)
main()
Mgf conversion loop examples¶
-
mgf_conversion_inside_and_outside_loop.main()¶
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import shutil
def main():
""""""
R = ursgal.UController(
profile="LTQ XL low res",
params={
"database": os.path.join(os.pardir, "example_data", "BSA.fasta"),
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation[]
],
},
)
engine = "omssa"
output_files = []
mzML_file = os.path.join(
os.pardir, "example_data", "mgf_conversion_example", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
R.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
R.params["http_output_folder"] = os.path.dirname(mzML_file)
R.fetch_file(engine="get_http_files_1_0_0")
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
# First method: Convert to MGF outside of the loop:
# (saves some time cause the MGF conversion is not always re-run)
mgf_file = R.convert(
input_file=mzML_file, engine="mzml2mgf_1_0_0" # from OpenMS example files
)
for prefix in ["10ppm", "20ppm"]:
R.params["prefix"] = prefix
output_file = R.search(
input_file=mgf_file,
engine=engine,
# output_file_name = 'some_userdefined_name'
)
# Second method: Automatically convert to MGF inside the loop:
# (MGF conversion is re-run every time because the prexix changed!)
for prefix in ["5ppm", "15ppm"]:
R.params["prefix"] = prefix
output_file = R.search(
input_file=mzML_file, # from OpenMS example files
engine=engine,
# output_file_name = 'another_fname',
)
output_files.append(output_file)
print("\tOutput files:")
for f in output_files:
print(f)
if __name__ == "__main__":
print(__doc__)
main()
Simple Venn diagram¶
-
simple_venn_example.main()¶ Example for plotting a simple Venn diagram with single ursgal csv files.
- usage:
- ./simple_venn_example.py
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Example for plotting a simple Venn diagram with single ursgal csv files.
usage:
./simple_venn_example.py
"""
uc = ursgal.UController(
profile="LTQ XL low res",
params={"visualization_label_positions": {"0": "omssa", "1": "xtandem"}},
)
file_list = [
os.path.join(
os.pardir, "tests", "data", "omssa_2_1_9", "test_BSA1_omssa_2_1_9.csv"
),
os.path.join(
os.pardir,
"tests",
"data",
"xtandem_sledgehammer",
"test_BSA1_xtandem_sledgehammer.csv",
),
]
uc.visualize(
input_files=file_list,
engine="venndiagram_1_1_0",
force=True,
)
return
if __name__ == "__main__":
main()
Sanitize csv¶
-
sanitize_combined_results.main(infile=None)¶ Sanitize an Ursgal result file after combining results from multiple search engines (or single search engine results, with modified parameters)
- usage:
- ./sanitize_combined_results.py <Ursgal_result_file>
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import sys
import os
def main(infile=None):
"""
Sanitize an Ursgal result file after combining results
from multiple search engines (or single search engine results,
with modified parameters)
usage:
./sanitize_combined_results.py <Ursgal_result_file>
"""
mass_spectrometer = "QExactive+"
params = {
"precursor_mass_tolerance_minus": 10,
"precursor_mass_tolerance_plus": 10,
"frag_mass_tolerance": 10,
"frag_mass_tolerance_unit": "ppm",
"-xmx": "32g",
}
uc = ursgal.UController(profile=mass_spectrometer, params=params)
# Parameters need to be adjusted based on the input file
# and the desired type of sanitizing.
# E.g. 'combined PEP' or 'Bayed PEP' can be used for combined PEP results,
# while 'PEP' should be used for results from single engines.
# A minimum difference between the top scoring, conflicting PSMs can be defined
# using the parameters 'score_diff_threshold' and 'threshold_is_log10'
uc.params.update(
{
"validation_score_field": "Bayes PEP",
# 'validation_score_field': 'PEP',
"bigger_scores_better": False,
"num_compared_psms": 25,
"accept_conflicting_psms": False,
"threshold_is_log10": True,
"score_diff_threshold": 0.0,
"psm_defining_colnames": [
"Spectrum Title",
"Sequence",
# 'Modifications',
# 'Charge',
# 'Is decoy',
],
"max_num_psms_per_spec": 1,
# 'preferred_engines': [],
"preferred_engines": [
"msfragger_2_3",
"pipi_1_4_6",
"moda_v1_61",
],
"remove_redundant_psms": False,
}
)
sanitized_combined_results = uc.execute_misc_engine(
input_file=infile,
engine="sanitize_csv",
)
if __name__ == "__main__":
main(
infile=sys.argv[1],
)
Test node execution¶
-
test_node_execution.main()¶ Testscript for executing the test node, which also tests the run time determination function.
- Usage:
- ./test_node_excution.py
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import os.path
import sys
import tempfile
import time
def main():
"""
Testscript for executing the test node, which also tests the run time
determination function.
Usage:
./test_node_excution.py
"""
uc = ursgal.UController(verbose=True)
temp_int, temp_fpath = tempfile.mkstemp(prefix="ursgal_", suffix=".csv")
temp_fobject = open(temp_fpath, "w")
print("test 1,2,3", file=temp_fobject)
temp_fobject.close()
test_1 = uc.execute_unode(temp_fpath, "_test_node")
test_2 = uc.execute_unode(test_1, "_test_node")
if __name__ == "__main__":
main()
Complete Workflow Scripts¶
Do it all folder wide¶
-
do_it_all_folder_wide.main(folder=None, profile=None, target_decoy_database=None)¶ An example test script to search all mzML files which are present in the specified folder. The search is currently performed on 4 search engines and 2 validation engines.
The machine profile has to be specified as well as the target-decoy database.
usage:
./do_it_all_folder_wide.py <mzML_folder> <profile> <target_decoy_database>Current profiles:
- ‘QExactive+’
- ‘LTQ XL low res’
- ‘LTQ XL high res’
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import sys
import glob
import os
def main(folder=None, profile=None, target_decoy_database=None):
"""
An example test script to search all mzML files which are present in the
specified folder. The search is currently performed on 4 search engines
and 2 validation engines.
The machine profile has to be specified as well as the target-decoy
database.
usage:
./do_it_all_folder_wide.py <mzML_folder> <profile> <target_decoy_database>
Current profiles:
* 'QExactive+'
* 'LTQ XL low res'
* 'LTQ XL high res'
"""
# define folder with mzML_files as sys.argv[1]
mzML_files = []
for mzml in glob.glob(os.path.join("{0}".format(folder), "*.mzML")):
mzML_files.append(mzml)
mass_spectrometer = profile
# We specify all search engines and validation engines that we want to use in a list
# (version numbers might differ on windows or mac):
search_engines = [
"omssa",
"xtandem_vengeance",
"msgfplus_v2016_09_16",
# 'msamanda_1_0_0_6300',
# 'myrimatch_2_1_138',
]
validation_engines = [
"percolator_2_08",
"qvality",
]
# Modifications that should be included in the search
all_mods = [
"C,fix,any,Carbamidomethyl",
"M,opt,any,Oxidation",
# 'N,opt,any,Deamidated',
# 'Q,opt,any,Deamidated',
# 'E,opt,any,Methyl',
# 'K,opt,any,Methyl',
# 'R,opt,any,Methyl',
"*,opt,Prot-N-term,Acetyl",
# 'S,opt,any,Phospho',
# 'T,opt,any,Phospho',
# 'N,opt,any,HexNAc'
]
# Initializing the Ursgal UController class with
# our specified modifications and mass spectrometer
params = {
"database": target_decoy_database,
"modifications": all_mods,
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
],
}
uc = ursgal.UController(profile=mass_spectrometer, params=params)
# complete workflow:
# every spectrum file is searched with every search engine,
# results are validated (for each engine seperately),
# validated results are merged and filtered for targets and PEP <= 0.01.
# In the end, all filtered results from all spectrum files are merged
for validation_engine in validation_engines:
result_files = []
for spec_file in mzML_files:
validated_results = []
for search_engine in search_engines:
unified_search_results = uc.search(
input_file=spec_file,
engine=search_engine,
)
validated_csv = uc.validate(
input_file=unified_search_results,
engine=validation_engine,
)
validated_results.append(validated_csv)
validated_results_from_all_engines = uc.execute_misc_engine(
input_file=validated_results,
engine="merge_csvs_1_0_0",
)
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_results_from_all_engines,
engine="filter_csv_1_0_0",
)
result_files.append(filtered_validated_results)
results_all_files = uc.execute_misc_engine(
input_file=result_files,
engine="merge_csvs_1_0_0",
)
if __name__ == "__main__":
if len(sys.argv) < 3:
print(main.__doc__)
sys.exit(1)
main(
folder=sys.argv[1],
profile=sys.argv[2],
target_decoy_database=sys.argv[3],
)
Large scale data analysis¶
-
barth_et_al_large_scale.main(folder)¶ Example script for reproducing the data for figure 3
usage:
./barth_et_al_large_scale.py <folder>The folder determines the target folder where the files will be downloaded
Chlamydomonas reinhardtii samples
Three biological replicates of 4 conditions (2_3, 2_4, 3_1, 4_1)
For more details on the samples please refer to Barth, J.; Bergner, S. V.; Jaeger, D.; Niehues, A.; Schulze, S.; Scholz, M.; Fufezan, C. The interplay of light and oxygen in the reactive oxygen stress response of Chlamydomonas reinhardtii dissected by quantitative mass spectrometry. MCP 2014, 13 (4), 969–989.
Merge all search results (per biological replicate and condition, on folder level) on engine level and validate via percolator.
‘LTQ XL high res’:
- repetition 1
- repetition 2
‘LTQ XL low res’:
- repetition 3
Database:
- Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta
Note
The database and the files will be automatically downloaded from our webpage and peptideatlas
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import os.path
import sys
def main(folder):
"""
Example script for reproducing the data for figure 3
usage:
./barth_et_al_large_scale.py <folder>
The folder determines the target folder where the files will be downloaded
Chlamydomonas reinhardtii samples
Three biological replicates of 4 conditions (2_3, 2_4, 3_1, 4_1)
For more details on the samples please refer to
Barth, J.; Bergner, S. V.; Jaeger, D.; Niehues, A.; Schulze, S.; Scholz,
M.; Fufezan, C. The interplay of light and oxygen in the reactive oxygen
stress response of Chlamydomonas reinhardtii dissected by quantitative mass
spectrometry. MCP 2014, 13 (4), 969–989.
Merge all search results (per biological replicate and condition, on folder
level) on engine level and validate via percolator.
'LTQ XL high res':
* repetition 1
* repetition 2
'LTQ XL low res':
* repetition 3
Database:
* Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta
Note:
The database and the files will be automatically downloaded from our
webpage and peptideatlas
"""
input_params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"modifications": [
"M,opt,any,Oxidation",
"*,opt,Prot-N-term,Acetyl", # N-Acetylation
],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_output_folder_root": folder,
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
uc = ursgal.UController(params=input_params)
if os.path.exists(input_params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
output_folder_to_file_list = {
("rep1_sample_2_3", "LTQ XL high res"): [
"CF_07062012_pH8_2_3A.mzML",
"CF_13062012_pH3_2_3A.mzML",
"CF_13062012_pH4_2_3A.mzML",
"CF_13062012_pH5_2_3A.mzML",
"CF_13062012_pH6_2_3A.mzML",
"CF_13062012_pH11FT_2_3A.mzML",
],
("rep1_sample_2_4", "LTQ XL high res"): [
"CF_07062012_pH8_2_4A.mzML",
"CF_13062012_pH3_2_4A_120615113039.mzML",
"CF_13062012_pH4_2_4A.mzML",
"CF_13062012_pH5_2_4A.mzML",
"CF_13062012_pH6_2_4A.mzML",
"CF_13062012_pH11FT_2_4A.mzML",
],
("rep1_sample_3_1", "LTQ XL high res"): [
"CF_12062012_pH8_1_3A.mzML",
"CF_13062012_pH3_1_3A.mzML",
"CF_13062012_pH4_1_3A.mzML",
"CF_13062012_pH5_1_3A.mzML",
"CF_13062012_pH6_1_3A.mzML",
"CF_13062012_pH11FT_1_3A.mzML",
],
("rep1_sample_4_1", "LTQ XL high res"): [
"CF_07062012_pH8_1_4A.mzML",
"CF_13062012_pH3_1_4A.mzML",
"CF_13062012_pH4_1_4A.mzML",
"CF_13062012_pH5_1_4A.mzML",
"CF_13062012_pH6_1_4A.mzML",
"CF_13062012_pH11FT_1_4A.mzML",
],
("rep2_sample_2_3", "LTQ XL high res"): [
"JB_18072012_2-3_A_FT.mzML",
"JB_18072012_2-3_A_pH3.mzML",
"JB_18072012_2-3_A_pH4.mzML",
"JB_18072012_2-3_A_pH5.mzML",
"JB_18072012_2-3_A_pH6.mzML",
"JB_18072012_2-3_A_pH8.mzML",
],
("rep2_sample_2_4", "LTQ XL high res"): [
"JB_18072012_2-4_A_FT.mzML",
"JB_18072012_2-4_A_pH3.mzML",
"JB_18072012_2-4_A_pH4.mzML",
"JB_18072012_2-4_A_pH5.mzML",
"JB_18072012_2-4_A_pH6.mzML",
"JB_18072012_2-4_A_pH8.mzML",
],
("rep2_sample_3_1", "LTQ XL high res"): [
"JB_18072012_3-1_A_FT.mzML",
"JB_18072012_3-1_A_pH3.mzML",
"JB_18072012_3-1_A_pH4.mzML",
"JB_18072012_3-1_A_pH5.mzML",
"JB_18072012_3-1_A_pH6.mzML",
"JB_18072012_3-1_A_pH8.mzML",
],
("rep2_sample_4_1", "LTQ XL high res"): [
"JB_18072012_4-1_A_FT.mzML",
"JB_18072012_4-1_A_pH3.mzML",
"JB_18072012_4-1_A_pH4.mzML",
"JB_18072012_4-1_A_pH5.mzML",
"JB_18072012_4-1_A_pH6.mzML",
"JB_18072012_4-1_A_pH8.mzML",
],
("rep3_sample_2_3", "LTQ XL low res"): [
"JB_FASP_pH3_2-3_28122012.mzML",
"JB_FASP_pH4_2-3_28122012.mzML",
"JB_FASP_pH5_2-3_28122012.mzML",
"JB_FASP_pH6_2-3_28122012.mzML",
"JB_FASP_pH8_2-3_28122012.mzML",
"JB_FASP_pH11-FT_2-3_28122012.mzML",
],
("rep3_sample_2_4", "LTQ XL low res"): [
"JB_FASP_pH3_2-4_28122012.mzML",
"JB_FASP_pH4_2-4_28122012.mzML",
"JB_FASP_pH5_2-4_28122012.mzML",
"JB_FASP_pH6_2-4_28122012.mzML",
"JB_FASP_pH8_2-4_28122012.mzML",
"JB_FASP_pH11-FT_2-4_28122012.mzML",
],
("rep3_sample_3_1", "LTQ XL low res"): [
"JB_FASP_pH3_3-1_28122012.mzML",
"JB_FASP_pH4_3-1_28122012.mzML",
"JB_FASP_pH5_3-1_28122012.mzML",
"JB_FASP_pH6_3-1_28122012.mzML",
"JB_FASP_pH8_3-1_28122012.mzML",
"JB_FASP_pH11-FT_3-1_28122012.mzML",
],
("rep3_sample_4_1", "LTQ XL low res"): [
"JB_FASP_pH3_4-1_28122012.mzML",
"JB_FASP_pH4_4-1_28122012.mzML",
"JB_FASP_pH5_4-1_28122012.mzML",
"JB_FASP_pH6_4-1_28122012.mzML",
"JB_FASP_pH8_4-1_28122012.mzML",
"JB_FASP_pH11-FT_4-1_28122012_130121201449.mzML",
],
}
for (outfolder, profile), mzML_file_list in sorted(
output_folder_to_file_list.items()
):
uc.params["ftp_output_folder"] = os.path.join(
input_params["ftp_output_folder_root"], outfolder
)
uc.params["ftp_include_ext"] = mzML_file_list
if os.path.exists(uc.params["ftp_output_folder"]) is False:
os.makedirs(uc.params["ftp_output_folder"])
uc.fetch_file(engine="get_ftp_files_1_0_0")
if os.path.exists(input_params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
search_engines = [
"omssa_2_1_9",
"xtandem_piledriver",
"myrimatch_2_1_138",
"msgfplus_v9979",
"msamanda_1_0_0_5243",
]
# This dict will be populated with the percolator-validated results
# of each engine ( 3 replicates x4 conditions = 12 files each )
percolator_results = {
"omssa_2_1_9": [],
"xtandem_piledriver": [],
"msgfplus_v9979": [],
"myrimatch_2_1_138": [],
"msamanda_1_0_0_5243": [],
}
five_files_for_venn_diagram = []
for search_engine in search_engines:
# This list will collect all 12 result files for each engine,
# after Percolator validation and filtering for PSMs with a
# FDR <= 0.01
filtered_results_of_engine = []
for mzML_dir_ext, mass_spectrometer in output_folder_to_file_list.keys():
# for mass_spectrometer, replicate_dir in replicates:
# for condition_dir in conditions:
uc.set_profile(mass_spectrometer)
mzML_dir = os.path.join(
input_params["ftp_output_folder_root"], mzML_dir_ext
)
# i.e. /media/plan-f/mzML/Christian_Fufezan/ROS_Experiment_2012/Juni_2012/2_3/Tech_A/
# all files ending with .mzml in that directory will be used!
unified_results_list = []
for filename in glob.glob(os.path.join(mzML_dir, "*.mzML")):
# print(filename)
if filename.lower().endswith(".mzml"):
# print(filename)
unified_search_results = uc.search(
input_file=filename,
engine=search_engine,
)
unified_results_list.append(unified_search_results)
# Merging results from the 6 pH-fractions:
merged_unified = uc.execute_misc_engine(
input_file=unified_results_list,
engine="merge_csvs_1_0_0",
)
# Validation with Percolator:
percolator_validated = uc.validate(
input_file=merged_unified,
engine="percolator_2_08", # one could replace this with 'qvality'
)
percolator_results[search_engine].append(percolator_validated)
# At this point, the analysis is finished. We got
# Percolator-validated results for each of the 3
# replicates and 12 conditions.
# But let's see how well the five search engines
# performed! To compare, we collect all PSMs with
# an estimated FDR <= 0.01 for each engine, and
# plot this information with the VennDiagram UNode.
# We will also use the Combine FDR Score method
# to combine the results from all five engines,
# and increase the number of identified peptides.
five_large_merged = []
filtered_final_results = []
# We will estimate the FDR for all 60 files
# (5 engines x12 files) when using percolator PEPs as
# quality score
uc.params["validation_score_field"] = "PEP"
uc.params["bigger_scores_better"] = False
# To make obtain smaller CSV files (and make plotting
# less RAM-intensive, we remove all decoys and PSMs above
# 0.06 FDR
uc.params["csv_filter_rules"] = [
["estimated_FDR", "lte", 0.06],
["Is decoy", "equals", "false"],
]
for engine, percolator_validated_list in percolator_results.items():
# unfiltered files for cFDR script
twelve_merged = uc.execute_misc_engine(
input_file=percolator_validated_list,
engine="merge_csvs_1_0_0",
)
twelve_filtered = []
for one_of_12 in percolator_validated_list:
one_of_12_FDR = uc.validate(
input_file=one_of_12, engine="add_estimated_fdr_1_0_0"
)
one_of_12_FDR_filtered = uc.execute_misc_engine(
input_file=one_of_12_FDR, engine="filter_csv_1_0_0"
)
twelve_filtered.append(one_of_12_FDR_filtered)
# For the combined FDR scoring, we merge all 12 files:
filtered_merged = uc.execute_misc_engine(
input_file=twelve_filtered, engine="merge_csvs_1_0_0"
)
five_large_merged.append(twelve_merged)
filtered_final_results.append(filtered_merged)
# The five big merged files of each engine are combined:
cFDR = uc.combine_search_results(
input_files=five_large_merged,
engine="combine_FDR_0_1",
)
# We estimate the FDR of this combined approach:
uc.params["validation_score_field"] = "Combined FDR Score"
uc.params["bigger_scores_better"] = False
cFDR_FDR = uc.validate(input_file=cFDR, engine="add_estimated_fdr_1_0_0")
# Removing decoys and low quality hits, to obtain a
# smaller file:
uc.params["csv_filter_rules"] = [
["estimated_FDR", "lte", 0.06],
["Is decoy", "equals", "false"],
]
cFDR_filtered_results = uc.execute_misc_engine(
input_file=cFDR_FDR,
engine="filter_csv_1_0_0",
)
filtered_final_results.append(cFDR_filtered_results)
# Since we produced quite a lot of files, let's print the full
# paths to our most important result files so we find them quickly:
print(
"""
These files can now be easily parsed and plotted with your
plotting tool of choice! We used the Python plotting library
matplotlib. Each unique combination of Sequence, modification
and charge was counted as a unique peptide.
"""
)
print("\n########### Result files: ##############")
for result_file in filtered_final_results:
print("\t*{0}".format(result_file))
if __name__ == "__main__":
if len(sys.argv) < 2:
print(main.__doc__)
sys.exit(1)
main(sys.argv[1])
Complete workflow for human BR dataset analysis¶
-
human_br_complete_workflow.main(folder)¶ usage:
./human_br_complete_workflow.py <folder_with_human_br_files>This scripts produces the data for figure 3.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
import pprint
def main(folder):
"""
usage:
./human_br_complete_workflow.py <folder_with_human_br_files>
This scripts produces the data for figure 3.
"""
# Initialize the UController:
uc = ursgal.UController(
params={
"enzyme": "trypsin",
"decoy_generation_mode": "reverse_protein",
}
)
# MS Spectra, downloaded from http://proteomecentral.proteomexchange.org
# via the dataset accession PXD000263 and converted to mzML
mass_spec_files = [
"120813OTc1_NQL-AU-0314-LFQ-LCM-SG-01_013.mzML",
"120813OTc1_NQL-AU-0314-LFQ-LCM-SG-02_025.mzML",
"120813OTc1_NQL-AU-0314-LFQ-LCM-SG-03_033.mzML",
"120813OTc1_NQL-AU-0314-LFQ-LCM-SG-04_048.mzML",
]
for mass_spec_file in mass_spec_files:
if os.path.exists(os.path.join(folder, mass_spec_file)) is False:
print(
"Please download RAW files to folder {} and convert to mzML:".format(
folder
)
)
pprint.pprint(mass_spec_files)
sys.exit(1)
# mods from Wen et al. (2015):
modifications = [
# Carbamidomethyl (C) was set as fixed modification
"C,fix,any,Carbamidomethyl",
"M,opt,any,Oxidation", # Oxidation (M) as well as
# Deamidated (NQ) were set as optional modification
"N,opt,any,Deamidated",
# Deamidated (NQ) were set as optional modification
"Q,opt,any,Deamidated",
]
# The target peptide database which will be searched (UniProt Human
# reference proteome from July 2013)
target_database = "uniprot_human_UP000005640_created_until_20130707.fasta"
# Let's turn it into a target decoy database by reversing peptides:
target_decoy_database = uc.execute_misc_engine(
input_file=target_database, engine="generate_target_decoy_1_0_0"
)
# OMSSA parameters from Wen et al. (2015):
omssa_params = {
# (used by default) # -w
"he": "1000", # -he 1000
"zcc": "1", # -zcc 1
"frag_mass_tolerance": "0.6", # -to 0.6
"frag_mass_tolerance_unit": "da", # -to 0.6
"precursor_mass_tolerance_minus": "10", # -te 10
"precursor_mass_tolerance_plus": "10", # -te 10
"precursor_mass_tolerance_unit": "ppm", # -teppm
"score_a_ions": False, # -i 1,4
"score_b_ions": True, # -i 1,4
"score_c_ions": False, # -i 1,4
"score_x_ions": False, # -i 1,4
"score_y_ions": True, # -i 1,4
"score_z_ions": False, # -i 1,4
"enzyme": "trypsin_p", # -e 10
"maximum_missed_cleavages": "1", # -v 1
"precursor_max_charge": "8", # -zh 8
"precursor_min_charge": "1", # -zl 1
"tez": "1", # -tez 1
"precursor_isotope_range": "0,1", # -ti 1
"num_match_spec": "1", # -hc 1
"database": target_decoy_database,
"modifications": modifications,
}
# MS-GF+ parameters from Wen et al. (2015):
msgf_params = {
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_unit": "ppm",
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_minus": "10",
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_plus": "10",
# the max number of optional modifications per peptide were set as 3
# (used by default) # number of allowed isotope errors was set as 1
"enzyme": "trypsin", # the enzyme was set as trypsin
# (used by default) # fully enzymatic peptides were specified, i.e. no non-enzymatic termini
"frag_method": "1", # the fragmentation method selected in the search was CID
"max_pep_length": "45", # the maximum peptide length to consider was set as 45
# the minimum precursor charge to consider if charges are not specified
# in the spectrum file was set as 1
"precursor_min_charge": "1",
# the maximum precursor charge to consider was set as 8
"precursor_max_charge": "8",
# (used by default) # the parameter 'addFeatures' was set as 1 (required for Percolator)
# all of the other parameters were set as default
# the instrument selected
# was High-res
"database": target_decoy_database,
"modifications": modifications,
}
# X!Tandem parameters from Wen et al. (2015):
xtandem_params = {
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_unit": "ppm",
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_minus": "10",
# precursor ion mass tolerance was set to 10 ppm
"precursor_mass_tolerance_plus": "10",
# the fragment ion mass tolerance was set to 0.6 Da
"frag_mass_tolerance": "0.6",
# the fragment ion mass tolerance was set to 0.6 Da
"frag_mass_tolerance_unit": "da",
# parent monoisotopic mass isotope error was set as 'yes'
"precursor_isotope_range": "0,1",
"precursor_max_charge": "8", # maximum parent charge of spectrum was set as 8
"enzyme": "trypsin", # the enzyme was set as trypsin ([RK]|[X])
# the maximum missed cleavage sites were set as 1
"maximum_missed_cleavages": "1",
# (used by default) # no model refinement was employed.
"database": target_decoy_database,
"modifications": modifications,
}
search_engine_settings = [
# not used in Wen et al., so we use the same settings as xtandem
("msamanda_1_0_0_5243", xtandem_params, "LTQ XL high res"),
# not used in Wen et al., so we use the same settings as xtandem
("myrimatch_2_1_138", xtandem_params, "LTQ XL high res"),
# the instrument selected was High-res
("msgfplus_v9979", msgf_params, "LTQ XL high res"),
("xtandem_jackhammer", xtandem_params, None),
("omssa_2_1_9", omssa_params, None),
]
merged_validated_files_3_engines = []
merged_validated_files_5_engines = []
for engine, wen_params, instrument in search_engine_settings:
# Initializing the uPLANIT UController class with
# our specified modifications and mass spectrometer
uc = ursgal.UController(params=wen_params)
if instrument is not None:
uc.set_profile(instrument)
unified_results = []
percolator_validated_results = []
for mzML_file in mass_spec_files:
unified_search_results = uc.search(
input_file=mzML_file,
engine=engine,
)
unified_results.append(unified_search_results)
validated_csv = uc.validate(
input_file=unified_search_results,
engine="percolator_2_08",
)
percolator_validated_results.append(validated_csv)
merged_validated_csv = uc.execute_misc_engine(
input_file=percolator_validated_results, engine="merge_csvs_1_0_0"
)
merged_unvalidated_csv = uc.execute_misc_engine(
input_file=unified_results,
engine="merge_csvs_1_0_0",
)
if engine in ["omssa_2_1_9", "xtandem_jackhammer", "msgfplus_v9979"]:
merged_validated_files_3_engines.append(merged_validated_csv)
merged_validated_files_5_engines.append(merged_validated_csv)
uc.params["prefix"] = "5-engines-summary"
uc.combine_search_results(
input_files=merged_validated_files_5_engines,
engine="combine_FDR_0_1",
)
uc.params["prefix"] = "3-engines-summary"
uc.combine_search_results(
input_files=merged_validated_files_3_engines,
engine="combine_FDR_0_1",
)
if __name__ == "__main__":
if len(sys.argv) < 2:
print(main.__doc__)
sys.exit(1)
main(sys.argv[1])
Complete workflow for analysis with pGlyco¶
-
example_pglyco_workflow.main(input_raw_file=None, database=None, enzyme=None)¶ - This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database, a database including only target sequences must be provided. In this database, the N-glycosylation motif N-X-S/T needs to be replaced with J-X-S/T, which will be done by the pGlyco wrapper.
- usage:
- ./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
#!/usr/bin/env python
# encoding: utf-8
import ursgal
import os
import sys
import shutil
def main(input_raw_file=None, database=None, enzyme=None):
"""
This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database,
a database including only target sequences must be provided.
In this database, the N-glycosylation motif N-X-S/T needs to be replaced
with J-X-S/T, which will be done by the pGlyco wrapper.
usage:
./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
"""
uc = ursgal.UController(
profile="QExactive+",
params={
"database": database,
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
"peptide_mapper_class_version": "UPeptideMapper_v4",
"enzyme": enzyme,
"frag_mass_tolerance": 20,
"frag_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
"aa_exception_dict": {},
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["q-value", "lte", 0.01],
["Conflicting uparam", "contains_not", "enzyme"],
],
},
)
xtracted_file = uc.convert(
input_file=input_raw_file,
engine="pparse_2_2_1",
# force = True,
)
mzml_file = uc.convert(
input_file=input_raw_file,
engine="thermo_raw_file_parser_1_1_2",
)
# os.remove(mzml_file.replace('.mzML', '.mgf'))
# os.remove(mzml_file.replace('.mzML', '.mgf.u.json'))
mgf_file = uc.convert(
input_file=mzml_file,
engine="mzml2mgf_2_0_0",
force=True,
)
search_result = uc.search_mgf(
input_file=xtracted_file,
engine="pglyco_db_2_2_2",
)
mapped_results = uc.execute_misc_engine(
input_file=search_result,
engine="upeptide_mapper",
)
unified_search_results = uc.execute_misc_engine(
input_file=mapped_results, engine="unify_csv"
)
validated_file = uc.validate(
input_file=search_result,
engine="pglyco_fdr_2_2_0",
)
mapped_validated_results = uc.execute_misc_engine(
input_file=validated_file,
engine="upeptide_mapper",
)
unified_validated_results = uc.execute_misc_engine(
input_file=mapped_validated_results, engine="unify_csv"
)
filtered_validated_results = uc.execute_misc_engine(
input_file=unified_validated_results,
engine="filter_csv",
)
return
if __name__ == "__main__":
main(input_raw_file=sys.argv[1], database=sys.argv[2], enzyme=sys.argv[3])
Complete workflow for analysis with TagGraph¶
-
example_pglyco_workflow.main(input_raw_file=None, database=None, enzyme=None) - This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database, a database including only target sequences must be provided. In this database, the N-glycosylation motif N-X-S/T needs to be replaced with J-X-S/T, which will be done by the pGlyco wrapper.
- usage:
- ./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
#!/usr/bin/env python
# encoding: utf-8
import ursgal
import os
import sys
import shutil
def main(input_raw_file=None, database=None, enzyme=None):
"""
This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database,
a database including only target sequences must be provided.
In this database, the N-glycosylation motif N-X-S/T needs to be replaced
with J-X-S/T, which will be done by the pGlyco wrapper.
usage:
./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
"""
uc = ursgal.UController(
profile="QExactive+",
params={
"database": database,
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
"peptide_mapper_class_version": "UPeptideMapper_v4",
"enzyme": enzyme,
"frag_mass_tolerance": 20,
"frag_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
"aa_exception_dict": {},
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["q-value", "lte", 0.01],
["Conflicting uparam", "contains_not", "enzyme"],
],
},
)
xtracted_file = uc.convert(
input_file=input_raw_file,
engine="pparse_2_2_1",
# force = True,
)
mzml_file = uc.convert(
input_file=input_raw_file,
engine="thermo_raw_file_parser_1_1_2",
)
# os.remove(mzml_file.replace('.mzML', '.mgf'))
# os.remove(mzml_file.replace('.mzML', '.mgf.u.json'))
mgf_file = uc.convert(
input_file=mzml_file,
engine="mzml2mgf_2_0_0",
force=True,
)
search_result = uc.search_mgf(
input_file=xtracted_file,
engine="pglyco_db_2_2_2",
)
mapped_results = uc.execute_misc_engine(
input_file=search_result,
engine="upeptide_mapper",
)
unified_search_results = uc.execute_misc_engine(
input_file=mapped_results, engine="unify_csv"
)
validated_file = uc.validate(
input_file=search_result,
engine="pglyco_fdr_2_2_0",
)
mapped_validated_results = uc.execute_misc_engine(
input_file=validated_file,
engine="upeptide_mapper",
)
unified_validated_results = uc.execute_misc_engine(
input_file=mapped_validated_results, engine="unify_csv"
)
filtered_validated_results = uc.execute_misc_engine(
input_file=unified_validated_results,
engine="filter_csv",
)
return
if __name__ == "__main__":
main(input_raw_file=sys.argv[1], database=sys.argv[2], enzyme=sys.argv[3])
Example search for 15N labeling and no label¶
-
search_with_label_15N.main()¶ Executes a search with 3 different search engines on an example file from the data from Barth et al. Two searches are performed pe engine for each 14N (unlabeled) and 15N labeling. The overlap of identified peptides for the 14N and 15N searches between the engines is visualized as well as the overlap between all 14N and 15N identified peptides.
- usage:
- ./search_with_label_15N.py
Note
It is important to convert the mgf file outside of (and before :) ) the search loop to avoid mgf file redundancy and to assure correct retention time mapping in the unify_csv node.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Executes a search with 3 different search engines on an example file from
the data from Barth et al. Two searches are performed pe engine for each
14N (unlabeled) and 15N labeling. The overlap of identified peptides
for the 14N and 15N searches between the engines is visualized as well as
the overlap between all 14N and 15N identified peptides.
usage:
./search_with_label_15N.py
Note:
It is important to convert the mgf file outside of (and before :) ) the
search loop to avoid mgf file redundancy and to assure correct retention
time mapping in the unify_csv node.
"""
engine_list = [
"omssa_2_1_9",
"xtandem_piledriver",
"msgfplus_v9979",
]
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"modifications": [],
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
],
"ftp_output_folder": os.path.join(
os.pardir, "example_data", "search_with_label_15N"
),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
if os.path.exists(params["ftp_output_folder"]) is False:
os.mkdir(params["ftp_output_folder"])
uc = ursgal.UController(profile="LTQ XL low res", params=params)
mzML_file = os.path.join(params["ftp_output_folder"], params["ftp_include_ext"][0])
if os.path.exists(mzML_file) is False:
uc.fetch_file(engine="get_ftp_files_1_0_0")
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
mgf_file = uc.convert(
input_file=mzML_file,
engine="mzml2mgf_1_0_0",
)
files_2_merge = {}
label_list = ["14N", "15N"]
for label in label_list:
validated_and_filtered_files_list = []
uc.params["label"] = label
uc.params["prefix"] = label
for engine in engine_list:
search_result = uc.search_mgf(
input_file=mgf_file,
engine=engine,
)
converted_result = uc.convert(
input_file=search_result,
guess_engine=True,
)
mapped_results = uc.execute_misc_engine(
input_file=converted_result,
engine="upeptide_mapper",
)
unified_search_results = uc.execute_misc_engine(
input_file=mapped_results, engine="unify_csv"
)
validated_file = uc.validate(
input_file=unified_search_results,
engine="percolator_2_08",
)
filtered_file = uc.execute_misc_engine(
input_file=validated_file, engine="filter_csv"
)
validated_and_filtered_files_list.append(filtered_file)
files_2_merge[label] = validated_and_filtered_files_list
uc.visualize(
input_files=validated_and_filtered_files_list,
engine="venndiagram_1_1_0",
)
uc.params["prefix"] = None
uc.params["label"] = ""
uc.params["visualization_label_positions"] = {}
label_comparison_file_list = []
for n, label in enumerate(label_list):
uc.params["visualization_label_positions"][str(n)] = label
label_comparison_file_list.append(
uc.execute_misc_engine(input_file=files_2_merge[label], engine="merge_csvs")
)
uc.visualize(
input_files=label_comparison_file_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
main()
Open Modification Search Scripts¶
Open modification search with three engines and combined PEP¶
-
open_modification_search_incl_combined_pep.main(folder=None, database=None, enzyme=None)¶ Example workflow to perform a open modification search with three independent search engines across all mzML files of a given folder and to statistically post-process and combine the results of all searches.
- Usage:
- ./open_modification_search_incl_combined_pep.py <mzML_folder> <database> <enzyme>
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import sys
import glob
import os
import pprint
from collections import defaultdict
def main(folder=None, database=None, enzyme=None):
"""
Example workflow to perform a open modification search with three independent search engines
across all mzML files of a given folder and to statistically post-process and combine the
results of all searches.
Usage:
./open_modification_search_incl_combined_pep.py <mzML_folder> <database> <enzyme>
"""
# For this particular dataset, two enzymes were used, namely gluc and trypsin.
mzml_files = []
for mzml in glob.glob(os.path.join(folder, "*.mzML")):
mzml_files.append(mzml)
mass_spectrometer = "QExactive+"
validation_engine = "percolator_3_4_0"
search_engines = ["msfragger_2_3", "pipi_1_4_6", "moda_v1_61"]
params = {
"modifications": ["C,fix,any,Carbamidomethyl"],
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
],
"frag_mass_tolerance_unit": "ppm",
"frag_mass_tolerance": 20,
"precursor_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
"moda_high_res": False,
"max_mod_size": 4000,
"min_mod_size": -200,
"precursor_true_units": "ppm",
"precursor_true_tolerance": 5,
"percolator_post_processing": "mix-max",
"psm_defining_colnames": [
"Spectrum Title",
"Sequence",
"Modifications",
"Charge",
"Is decoy",
"Mass Difference",
],
"database": database,
"enzyme": enzyme,
}
uc = ursgal.UController(
profile=mass_spectrometer,
params=params,
)
# This will hold input to combined PEP engine
combined_pep_input = defaultdict(list)
# This dictionary will help organize which results to merge
all_merged_results = defaultdict(list)
for search_engine in search_engines:
# The modification size for MSFragger is configured through precursor mass tolerance
if search_engine == "msfragger_2_3":
uc.params.update(
{
"precursor_mass_tolerance_unit": "da",
"precursor_mass_tolerance_plus": 4000,
"precursor_mass_tolerance_minus": 200,
}
)
for n, spec_file in enumerate(mzml_files):
# 1. convert to MGF
mgf_file = uc.convert(
input_file=spec_file,
engine="mzml2mgf_2_0_0",
)
# 2. do the actual search
raw_search_results = uc.search_mgf(
input_file=mgf_file,
engine=search_engine,
)
# reset precursor mass tolerance just in case it was previously changed
uc.params.update(
{
"precursor_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
}
)
# 3. convert files to csv
csv_search_results = uc.convert(
input_file=raw_search_results,
engine=None,
guess_engine=True,
)
# 4. protein mapping.
mapped_csv_search_results = uc.execute_misc_engine(
input_file=csv_search_results,
engine="upeptide_mapper_1_0_0",
)
# 5. Convert csv to unified ursgal csv format:
unified_search_results = uc.execute_misc_engine(
input_file=mapped_csv_search_results,
engine="unify_csv_1_0_0",
merge_duplicates=False,
)
# 6. Validate the results
validated_csv = uc.validate(
input_file=unified_search_results,
engine=validation_engine,
)
# save the validated input for combined pep
# Eventually, each sample will have 3 files correpsonding to the 3 search engines
combined_pep_input["sample_{0}".format(n)].append(validated_csv)
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_csv,
engine="filter_csv_1_0_0",
merge_duplicates=False,
)
all_merged_results["percolator_only"].append(filtered_validated_results)
# combined pep
uc.params.update(
{
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["combined PEP", "lte", 0.01],
],
"psm_defining_colnames": [
"Spectrum Title",
"Sequence",
"Modifications",
"Charge",
"Is decoy",
],
}
)
for sample in combined_pep_input.keys():
combine_results = uc.execute_misc_engine(
input_file=combined_pep_input[sample],
engine="combine_pep_1_0_0",
)
filtered_validated_results = uc.execute_misc_engine(
input_file=combine_results,
engine="filter_csv_1_0_0",
)
all_merged_results["combined_pep"].append(filtered_validated_results)
# separately merge results from the two types of validation techniques
# We also add back "Mass Difference" to columns defining a PSM to avoid merging mass differences
uc.params.update(
{
"psm_defining_colnames": [
"Spectrum Title",
"Sequence",
"Modifications",
"Charge",
"Is decoy",
"Mass Difference",
],
}
)
for validation_type in all_merged_results.keys():
if validation_type == "percolator_only":
uc.params["psm_colnames_to_merge_multiple_values"] = {
"PEP": "min_value",
}
else:
uc.params["psm_colnames_to_merge_multiple_values"] = {
"combined PEP": "min_value",
"Bayes PEP": "min_value",
}
uc.params["prefix"] = "All_{0}".format(
validation_type
) # helps recognize files easily
merged_results_one_rep = uc.execute_misc_engine(
input_file=all_merged_results[validation_type],
engine="merge_csvs_1_0_0",
merge_duplicates=True,
)
uc.params["prefix"] = ""
if __name__ == "__main__":
main(
folder=sys.argv[1],
database=sys.argv[2],
enzyme=sys.argv[3],
)
Complete workflow for analysis with TagGraph¶
-
example_pglyco_workflow.main(input_raw_file=None, database=None, enzyme=None) - This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database, a database including only target sequences must be provided. In this database, the N-glycosylation motif N-X-S/T needs to be replaced with J-X-S/T, which will be done by the pGlyco wrapper.
- usage:
- ./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
#!/usr/bin/env python
# encoding: utf-8
import ursgal
import os
import sys
import shutil
def main(input_raw_file=None, database=None, enzyme=None):
"""
This script executes three steps:
- convert .raw file using pParse
- search the resulting .mgf file using pGlyco
- validate results using pGlycoFDR
Since pGlyco does not support providing a custom target-decoy database,
a database including only target sequences must be provided.
In this database, the N-glycosylation motif N-X-S/T needs to be replaced
with J-X-S/T, which will be done by the pGlyco wrapper.
usage:
./simple_example_pglyco_workflow.py <input_raw_file> <database> <enzyme>
"""
uc = ursgal.UController(
profile="QExactive+",
params={
"database": database,
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
"peptide_mapper_class_version": "UPeptideMapper_v4",
"enzyme": enzyme,
"frag_mass_tolerance": 20,
"frag_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
"aa_exception_dict": {},
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["q-value", "lte", 0.01],
["Conflicting uparam", "contains_not", "enzyme"],
],
},
)
xtracted_file = uc.convert(
input_file=input_raw_file,
engine="pparse_2_2_1",
# force = True,
)
mzml_file = uc.convert(
input_file=input_raw_file,
engine="thermo_raw_file_parser_1_1_2",
)
# os.remove(mzml_file.replace('.mzML', '.mgf'))
# os.remove(mzml_file.replace('.mzML', '.mgf.u.json'))
mgf_file = uc.convert(
input_file=mzml_file,
engine="mzml2mgf_2_0_0",
force=True,
)
search_result = uc.search_mgf(
input_file=xtracted_file,
engine="pglyco_db_2_2_2",
)
mapped_results = uc.execute_misc_engine(
input_file=search_result,
engine="upeptide_mapper",
)
unified_search_results = uc.execute_misc_engine(
input_file=mapped_results, engine="unify_csv"
)
validated_file = uc.validate(
input_file=search_result,
engine="pglyco_fdr_2_2_0",
)
mapped_validated_results = uc.execute_misc_engine(
input_file=validated_file,
engine="upeptide_mapper",
)
unified_validated_results = uc.execute_misc_engine(
input_file=mapped_validated_results, engine="unify_csv"
)
filtered_validated_results = uc.execute_misc_engine(
input_file=unified_validated_results,
engine="filter_csv",
)
return
if __name__ == "__main__":
main(input_raw_file=sys.argv[1], database=sys.argv[2], enzyme=sys.argv[3])
Processing of mass differences using PTM-Shepherd¶
-
ptmshepherd_mass_difference_processing.main(folder=None, sanitized_search_results=None)¶ Downstream processing of mass differences from open modification search results using PTM-Shepherd. A folder containing all relevant mzML files and a sanitized (one PSM per spectrum) Ursgal result file (containing open modification search results) are required.
- usage:
- ./ptmshepherd_mass_difference_processing.py <folder_with_mzML> <sanitized_result_file>
#!/usr/bin/env python3
import os
import ursgal
import sys
import glob
from collections import defaultdict as ddict
import csv
def main(folder=None, sanitized_search_results=None):
"""
Downstream processing of mass differences from open modification search results
using PTM-Shepherd.
A folder containing all relevant mzML files and a sanitized (one PSM per spectrum)
Ursgal result file (containing open modification search results) are required.
usage:
./ptmshepherd_mass_difference_processing.py <folder_with_mzML> <sanitized_result_file>
"""
mzml_files = []
for mzml in glob.glob(os.path.join(folder, "*.mzML")):
mzml_files.append(mzml)
params = {
"use_pyqms_for_mz_calculation": True,
"cpus": 8,
"precursor_mass_tolerance_unit": "ppm",
"precursor_mass_tolerance_plus": 5,
"precursor_mass_tolerance_minus": 5,
"frag_mass_tolerance_unit": "ppm",
"frag_mass_tolerance": 20,
"modifications": [
"C,opt,any,Carbamidomethyl",
],
"-xmx": "12G",
"psm_defining_colnames": [
"Spectrum Title",
"Sequence",
# 'Modifications',
# 'Mass Difference',
# 'Charge',
# 'Is decoy',
],
"mzml_input_files": mzml_files,
"validation_score_field": "combined PEP",
"bigger_scores_better": False,
}
uc = ursgal.UController(params=params, profile="QExactive+", verbose=False)
ptmshepherd_results = uc.validate(
input_file=sanitized_search_results,
engine="ptmshepherd_0_3_5",
# force=True,
)
if __name__ == "__main__":
main(
folder=sys.argv[1],
sanitized_search_results=sys.argv[2],
)
Version Comparison Scripts¶
X!Tandem version comparison¶
-
xtandem_version_comparison.main()¶ Executes a search with 5 versions of X!Tandem on an example file from the data from Barth et al. 2014.
- usage:
- ./xtandem_version_comparison.py
This is a simple example file to show the straightforward comparison of different program versions of X!Tandem
Creates a Venn diagram with the peptides obtained by the different versions.
Note
At the moment in total 6 XTandem versions are incorporated in Ursgal.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Executes a search with 5 versions of X!Tandem on an example file from the
data from Barth et al. 2014.
usage:
./xtandem_version_comparison.py
This is a simple example file to show the straightforward comparison of
different program versions of X!Tandem
Creates a Venn diagram with the peptides obtained by the different versions.
Note:
At the moment in total 6 XTandem versions are incorporated in Ursgal.
"""
engine_list = [
# 'xtandem_cyclone',
"xtandem_jackhammer",
"xtandem_sledgehammer",
"xtandem_piledriver",
"xtandem_vengeance",
"xtandem_alanine",
]
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"modifications": [],
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
],
"ftp_output_folder": os.path.join(
os.pardir, "example_data", "xtandem_version_comparison"
),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
if os.path.exists(params["ftp_output_folder"]) is False:
os.mkdir(params["ftp_output_folder"])
uc = ursgal.UController(profile="LTQ XL low res", params=params)
mzML_file = os.path.join(params["ftp_output_folder"], params["ftp_include_ext"][0])
if os.path.exists(mzML_file) is False:
uc.fetch_file(engine="get_ftp_files_1_0_0")
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
filtered_files_list = []
for engine in engine_list:
unified_result_file = uc.search(
input_file=mzML_file,
engine=engine,
)
validated_file = uc.validate(
input_file=unified_result_file,
engine="percolator_2_08",
)
filtered_file = uc.execute_misc_engine(
input_file=validated_file,
engine="filter_csv_1_0_0",
)
filtered_files_list.append(filtered_file)
uc.visualize(
input_files=filtered_files_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
main()
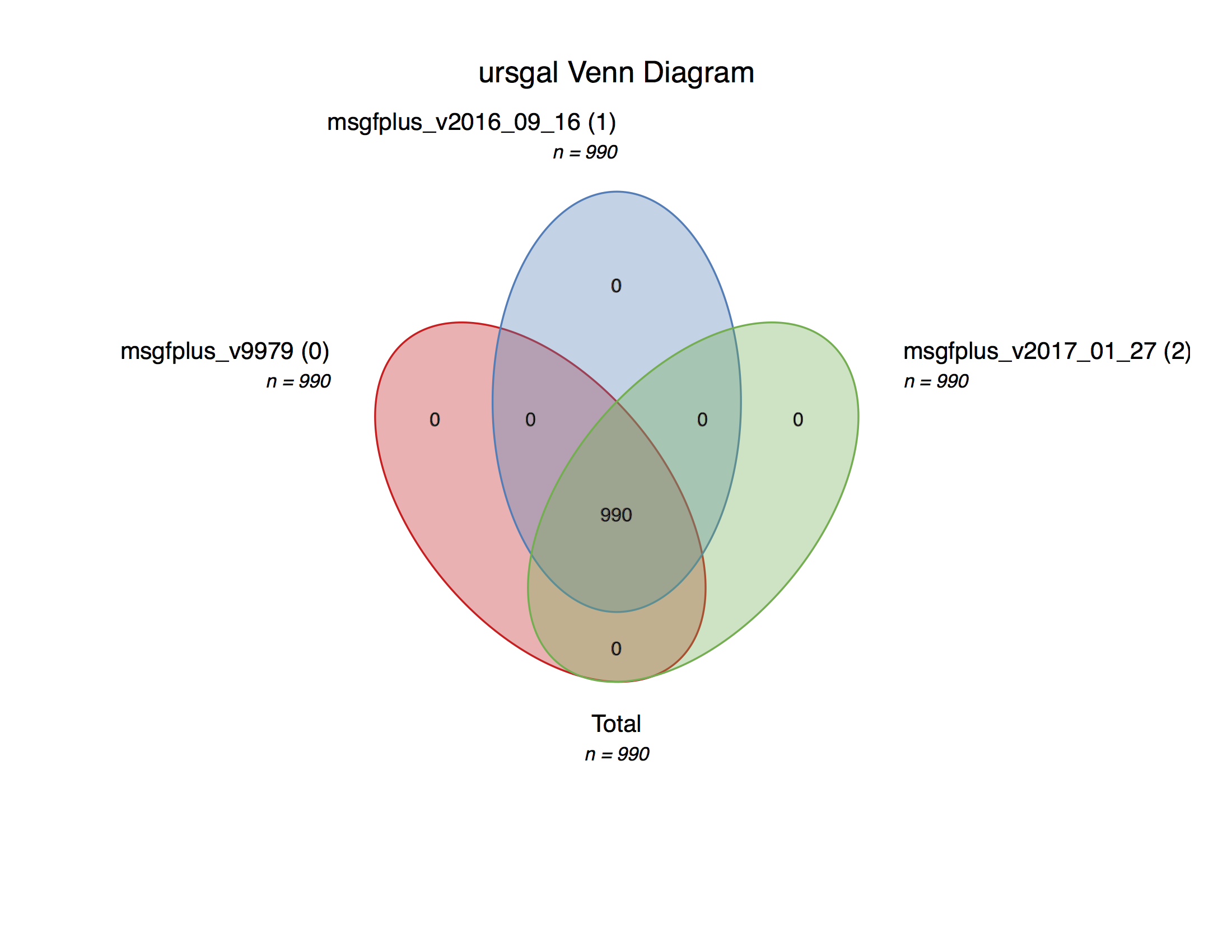
MSGF+ version comparison¶
-
msgf_version_comparison.main()¶ Executes a search with a current maximum of 3 versions of MSGF+ on an example file from the data from Barth et al. (2014)
- usage:
- ./msgf_version_comparison.py
This is a simple example file to show the straightforward comparison of different program versions of MSGF+
Creates a Venn diagram with the peptides obtained by the different versions.
An example plot can be found in the online documentation.
Note
Uses the new MS-GF+ C# mzid converter if available
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Executes a search with a current maximum of 3 versions of MSGF+ on an
example file from the data from Barth et al. (2014)
usage:
./msgf_version_comparison.py
This is a simple example file to show the straightforward comparison of
different program versions of MSGF+
Creates a Venn diagram with the peptides obtained by the different
versions.
An example plot can be found in the online documentation.
Note:
Uses the new MS-GF+ C# mzid converter if available
"""
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]],
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
],
"ftp_output_folder": os.path.join(
os.pardir, "example_data", "msgf_version_comparison"
),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
"remove_temporary_files": False,
}
if os.path.exists(params["ftp_output_folder"]) is False:
os.mkdir(params["ftp_output_folder"])
uc = ursgal.UController(profile="LTQ XL low res", params=params)
engine_list = [
"msgfplus_v9979",
"msgfplus_v2016_09_16",
]
if "msgfplus_v2017_01_27" in uc.unodes.keys():
if uc.unodes["msgfplus_v2017_01_27"]["available"]:
engine_list.append("msgfplus_v2017_01_27")
if "msgfplus_v2018_01_30" in uc.unodes.keys():
if uc.unodes["msgfplus_v2018_01_30"]["available"]:
engine_list.append("msgfplus_v2018_01_30")
if "msgfplus_C_mzid2csv_v2017_07_04" in uc.unodes.keys():
if uc.unodes["msgfplus_C_mzid2csv_v2017_07_04"]["available"]:
uc.params[
"msgfplus_mzid_converter_version"
] = "msgfplus_C_mzid2csv_v2017_07_04"
mzML_file = os.path.join(params["ftp_output_folder"], params["ftp_include_ext"][0])
if os.path.exists(mzML_file) is False:
uc.fetch_file(engine="get_ftp_files_1_0_0")
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
filtered_files_list = []
for engine in engine_list:
unified_result_file = uc.search(
input_file=mzML_file,
engine=engine,
)
validated_file = uc.validate(
input_file=unified_result_file,
engine="percolator_2_08",
)
filtered_file = uc.execute_misc_engine(
input_file=validated_file,
engine="filter_csv_1_0_0",
)
filtered_files_list.append(filtered_file)
uc.visualize(
input_files=filtered_files_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
main()
Bruderer et al. (2015) X!Tandem comparison incl. machine ppm offset¶
-
xtandem_version_comparison_qexactive.main(folder)¶ Executes a search with 5 versions of X!Tandem on an example file from the data from Bruderer et al. 2015.
- usage:
- ./xtandem_version_comparison.py <folder containing B_D140314_SGSDSsample1_R01_MSG_T0.mzML>
This is a simple example file to show the straightforward comparison of different program versions of X!Tandem, similar to the example script ‘xtandem_version_comparison’, but analyzing high resolution data which can be better handled by version newer than Jackhammer. One gains approximately 10 percent more peptides with newer versions of X!Tandem.
Creates a Venn diagram with the peptides obtained by the different versions.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
import sys
def main(folder):
"""
Executes a search with 5 versions of X!Tandem on an example file from the
data from Bruderer et al. 2015.
usage:
./xtandem_version_comparison.py <folder containing B_D140314_SGSDSsample1_R01_MSG_T0.mzML>
This is a simple example file to show the straightforward comparison of
different program versions of X!Tandem, similar to the example script
'xtandem_version_comparison', but analyzing high resolution data
which can be better handled by version newer than Jackhammer. One gains
approximately 10 percent more peptides with newer versions of X!Tandem.
Creates a Venn diagram with the peptides obtained by the different versions.
"""
required_example_file = "B_D140314_SGSDSsample1_R01_MSG_T0.mzML"
if os.path.exists(os.path.join(folder, required_example_file)) is False:
print(
"""
Your specified folder does not contain the required example file:
{0}
The RAW data from peptideatlas.org (PASS00589, password: WF6554orn)
will be downloaded.
Please convert to mzML after the download has finished and run this
script again.
""".format(
required_example_file
)
)
ftp_get_params = {
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00589",
"ftp_password": "WF6554orn",
"ftp_include_ext": [required_example_file.replace(".mzML", ".raw")],
"ftp_output_folder": folder,
}
uc = ursgal.UController(params=ftp_get_params)
uc.fetch_file(engine="get_ftp_files_1_0_0")
sys.exit(1)
engine_list = [
"xtandem_cyclone",
"xtandem_jackhammer",
"xtandem_sledgehammer",
"xtandem_piledriver",
"xtandem_vengeance",
]
params = {
"database": os.path.join(
os.pardir, "example_data", "hs_201303_qs_sip_target_decoy.fasta"
),
"modifications": ["C,fix,any,Carbamidomethyl"],
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]],
"http_url": "http://www.uni-muenster.de/Biologie.IBBP.AGFufezan/misc/hs_201303_qs_sip_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
"machine_offset_in_ppm": -5e-6,
}
uc = ursgal.UController(profile="QExactive+", params=params)
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
mzML_file = os.path.join(folder, required_example_file)
filtered_files_list = []
for engine in engine_list:
unified_result_file = uc.search(
input_file=mzML_file,
engine=engine,
force=False,
)
validated_file = uc.validate(
input_file=unified_result_file,
engine="percolator_2_08",
)
filtered_file = uc.execute_misc_engine(
input_file=validated_file,
engine="filter_csv_1_0_0",
)
filtered_files_list.append(filtered_file)
uc.visualize(
input_files=filtered_files_list,
engine="venndiagram_1_1_0",
)
return
if __name__ == "__main__":
if len(sys.argv) < 2:
print(main.__doc__)
sys.exit(1)
main(sys.argv[1])
Parameter Optimization Scripts¶
BSA machine ppm offset example¶
-
bsa_ppm_offset_test.main()¶ Example script to do a simple machine ppm offset parameter sweep. The m/z values in the example mgf file are stepwise changed and the in the final output the total peptides are counted.
- usage:
- ./bsa_ppm_offset_test.py
Note
As expected, if the offset becomes to big no peptides can be found anymore.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import csv
from collections import defaultdict as ddict
import os
import shutil
def main():
"""
Example script to do a simple machine ppm offset parameter sweep.
The m/z values in the example mgf file are stepwise changed and the in the
final output the total peptides are counted.
usage:
./bsa_ppm_offset_test.py
Note:
As expected, if the offset becomes to big no peptides can be found anymore.
"""
ppm_offsets = [
(-10, "-10_ppm_offset"),
(-9, "-9_ppm_offset"),
(-8, "-8_ppm_offset"),
(-7, "-7_ppm_offset"),
(-6, "-6_ppm_offset"),
(-5, "-5_ppm_offset"),
(-4, "-4_ppm_offset"),
(-3, "-3_ppm_offset"),
(-2, "-2_ppm_offset"),
(-1, "-1_ppm_offset"),
(None, "0_ppm_offset"),
(1, "1_ppm_offset"),
(2, "2_ppm_offset"),
(3, "3_ppm_offset"),
(4, "4_ppm_offset"),
(5, "5_ppm_offset"),
(6, "6_ppm_offset"),
(7, "7_ppm_offset"),
(8, "8_ppm_offset"),
(9, "9_ppm_offset"),
(10, "10_ppm_offset"),
]
engine_list = ["xtandem_vengeance"]
R = ursgal.UController(
profile="LTQ XL low res",
params={
"database": os.path.join(os.pardir, "example_data", "BSA.fasta"),
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
},
)
mzML_file = os.path.join(
os.pardir, "example_data", "BSA_machine_ppm_offset_example", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
R.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
R.params["http_output_folder"] = os.path.dirname(mzML_file)
R.fetch_file(engine="get_http_files_1_0_0")
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
for engine in engine_list:
for (ppm_offset, prefix) in ppm_offsets:
R.params["machine_offset_in_ppm"] = ppm_offset
R.params["prefix"] = prefix
unified_search_result_file = R.search(
input_file=mzML_file,
engine=engine,
force=False,
)
collector = ddict(set)
for csv_path in glob.glob("{0}/*/*unified.csv".format(os.path.dirname(mzML_file))):
for line_dict in csv.DictReader(open(csv_path, "r")):
collector[csv_path].add(line_dict["Sequence"])
for csv_path, peptide_set in sorted(collector.items()):
file_name = os.path.basename(csv_path)
offset = file_name.split("_")[0]
print(
"Search with {0: >3} ppm offset found {1: >2} peptides".format(
offset, len(peptide_set)
)
)
return
if __name__ == "__main__":
main()
Precursor mass tolerance example¶
-
bsa_precursor_mass_tolerance_example.main()¶ Example script to do a precursor mass tolerance parameter sweep.
- usage:
- ./bsa_precursor_mass_tolerance_example.py
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import csv
from collections import defaultdict as ddict
import os
import shutil
def main():
"""
Example script to do a precursor mass tolerance parameter sweep.
usage:
./bsa_precursor_mass_tolerance_example.py
"""
precursor_mass_tolerance_list = [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
]
engine_list = ["xtandem_vengeance"]
R = ursgal.UController(
profile="LTQ XL low res",
params={
"database": os.path.join(os.pardir, "example_data", "BSA.fasta"),
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
},
)
mzML_file = os.path.join(
os.pardir, "example_data", "BSA_precursor_mass_tolerance_example", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
R.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
R.params["http_output_folder"] = os.path.dirname(mzML_file)
R.fetch_file(engine="get_http_files_1_0_0")
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
# Convert mzML to MGF outside the loop, so this step is not repeated in
# the loop
mgf_file = R.convert(input_file=mzML_file, engine="mzml2mgf_1_0_0")
for engine in engine_list:
for precursor_mass_tolerance in precursor_mass_tolerance_list:
R.params["precursor_mass_tolerance_minus"] = precursor_mass_tolerance
R.params["precursor_mass_tolerance_plus"] = precursor_mass_tolerance
R.params["prefix"] = "{0}_precursor_mass_tolerance_".format(
precursor_mass_tolerance
)
unified_search_result_file = R.search(
input_file=mgf_file,
engine=engine,
force=False,
)
collector = ddict(set)
for csv_path in glob.glob("{0}/*/*unified.csv".format(os.path.dirname(mzML_file))):
for line_dict in csv.DictReader(open(csv_path, "r")):
collector[csv_path].add(line_dict["Sequence"])
for csv_path, peptide_set in sorted(collector.items()):
file_name = os.path.basename(csv_path)
tolerance = file_name.split("_")[0]
print(
"Search with {0: >2} ppm precursor mass tolerance found {1: >2} peptides".format(
tolerance, len(peptide_set)
)
)
return
if __name__ == "__main__":
main()
Fragment mass tolerance example¶
-
bsa_fragment_mass_tolerance_example.main()¶ Example script to do a fragment mass tolerance parameter sweep.
- usage:
- ./bsa_fragment_mass_tolerance_example.py
If the fragment mass tolerance becomes to small, ver few peptides are found. With this small sweep the actual min accuracy of a mass spectrometer can be estimated.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import csv
from collections import defaultdict as ddict
import os
import shutil
def main():
"""
Example script to do a fragment mass tolerance parameter sweep.
usage:
./bsa_fragment_mass_tolerance_example.py
If the fragment mass tolerance becomes to small, ver few peptides are found.
With this small sweep the actual min accuracy of a mass spectrometer can
be estimated.
"""
fragment_mass_tolerance_list = [
0.02,
0.04,
0.06,
0.08,
0.1,
0.2,
0.3,
0.4,
0.5,
]
engine_list = ["xtandem_vengeance"]
R = ursgal.UController(
profile="LTQ XL low res",
params={
"database": os.path.join(os.pardir, "example_data", "BSA.fasta"),
"modifications": [
"M,opt,any,Oxidation", # Met oxidation
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
"*,opt,Prot-N-term,Acetyl", # N-Acteylation
],
},
)
mzML_file = os.path.join(
os.pardir, "example_data", "BSA_fragment_mass_tolerance_example", "BSA1.mzML"
)
if os.path.exists(mzML_file) is False:
R.params[
"http_url"
] = "http://sourceforge.net/p/open-ms/code/HEAD/tree/OpenMS/share/OpenMS/examples/BSA/BSA1.mzML?format=raw"
R.params["http_output_folder"] = os.path.dirname(mzML_file)
R.fetch_file(engine="get_http_files_1_0_0")
try:
shutil.move("{0}?format=raw".format(mzML_file), mzML_file)
except:
shutil.move("{0}format=raw".format(mzML_file), mzML_file)
# Convert mzML to MGF outside the loop, so this step is not repeated in
# the loop
mgf_file = R.convert(
input_file=mzML_file,
engine="mzml2mgf_1_0_0",
)
for engine in engine_list:
for fragment_mass_tolerance in fragment_mass_tolerance_list:
R.params["frag_mass_tolerance"] = fragment_mass_tolerance
R.params["prefix"] = "{0}_fragment_mass_tolerance_".format(
fragment_mass_tolerance
)
unified_search_result_file = R.search(
input_file=mgf_file,
engine=engine,
force=False,
)
collector = ddict(set)
for csv_path in glob.glob("{0}/*/*unified.csv".format(os.path.dirname(mzML_file))):
for line_dict in csv.DictReader(open(csv_path, "r")):
collector[csv_path].add(line_dict["Sequence"])
for csv_path, peptide_set in sorted(collector.items()):
file_name = os.path.basename(csv_path)
tolerance = file_name.split("_")[0]
print(
"Search with {0: <4} Da fragment mass tolerance found {1: >2} peptides".format(
tolerance, len(peptide_set)
)
)
return
if __name__ == "__main__":
main()
Bruderer et al. (2015) machine offset sweep (data for figure 2)¶
-
machine_offset_bruderer_sweep.search(input_folder=None)¶ Does the parameter sweep on every tenth MS2 spectrum of the data from Bruderer et al. (2015) und X!Tandem Sledgehammer.
Note
Please download the .RAW data for the DDA dataset from peptideatlas.org (PASS00589, password: WF6554orn) and convert to mzML. Then the script can be executed with the folder with the mzML files as the first argument.
Warning
This script (if the sweep ranges are not changed) will perform 10080 searches which will produce approximately 100 GB output (inclusive mzML files)
usage:
./machine_offset_bruderer_sweep.py <folder_with_bruderer_data>Sweeps over:
- machine_offset_in_ppm from -20 to +20 ppm offset
- precursor mass tolerance from 1 to 5 ppm
- fragment mass tolerance from -2.5 to 20 ppm
The search can be very time consuming (depending on your machine/cluster), therefor the analyze step can be performed separately by calling analyze() instead of search() when one has already performed the searches and wants to analyze the results.
-
machine_offset_bruderer_sweep.analyze(folder)¶ Parses the result files form search and write a result .csv file which contains the data to plot figure 2.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import csv
import os
from collections import defaultdict as ddict
import sys
import re
MQ_OFFSET_TO_FILENAME = [
(4.71, "B_D140314_SGSDSsample2_R01_MSG_T0.mzML"),
(4.98, "B_D140314_SGSDSsample6_R01_MSG_T0.mzML"),
(4.9, "B_D140314_SGSDSsample1_R01_MSG_T0.mzML"),
(5.41, "B_D140314_SGSDSsample4_R01_MSG_T0.mzML"),
(5.78, "B_D140314_SGSDSsample5_R01_MSG_T0.mzML"),
(6.01, "B_D140314_SGSDSsample8_R01_MSG_T0.mzML"),
(6.22, "B_D140314_SGSDSsample7_R01_MSG_T0.mzML"),
(6.83, "B_D140314_SGSDSsample3_R01_MSG_T0.mzML"),
(7.61, "B_D140314_SGSDSsample4_R02_MSG_T0.mzML"),
(7.59, "B_D140314_SGSDSsample8_R02_MSG_T0.mzML"),
(7.93, "B_D140314_SGSDSsample6_R02_MSG_T0.mzML"),
(7.91, "B_D140314_SGSDSsample1_R02_MSG_T0.mzML"),
(8.23, "B_D140314_SGSDSsample3_R02_MSG_T0.mzML"),
(8.33, "B_D140314_SGSDSsample7_R02_MSG_T0.mzML"),
(9.2, "B_D140314_SGSDSsample5_R02_MSG_T0.mzML"),
(9.4, "B_D140314_SGSDSsample2_R02_MSG_T0.mzML"),
(9.79, "B_D140314_SGSDSsample1_R03_MSG_T0.mzML"),
(10.01, "B_D140314_SGSDSsample3_R03_MSG_T0.mzML"),
(10.03, "B_D140314_SGSDSsample7_R03_MSG_T0.mzML"),
(10.58, "B_D140314_SGSDSsample2_R03_MSG_T0.mzML"),
(11.1, "B_D140314_SGSDSsample4_R03_MSG_T0.mzML"),
(11.21, "B_D140314_SGSDSsample5_R03_MSG_T0.mzML"),
(11.45, "B_D140314_SGSDSsample6_R03_MSG_T0.mzML"),
(12.19, "B_D140314_SGSDSsample8_R03_MSG_T0.mzML"),
]
GENERAL_PARAMS = {
"database": os.path.join(
os.pardir, "example_data", "hs_201303_qs_sip_target_decoy.fasta"
),
"modifications": [
"C,fix,any,Carbamidomethyl", # Carbamidomethylation
],
"scan_skip_modulo_step": 10,
"http_url": "http://www.uni-muenster.de/Biologie.IBBP.AGFufezan/misc/hs_201303_qs_sip_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
def search(input_folder=None):
"""
Does the parameter sweep on every tenth MS2 spectrum of the data from
Bruderer et al. (2015) und X!Tandem Sledgehammer.
Note:
Please download the .RAW data for the DDA dataset from peptideatlas.org
(PASS00589, password: WF6554orn) and convert to mzML.
Then the script can be executed with the folder with the mzML files as
the first argument.
Warning:
This script (if the sweep ranges are not changed) will perform 10080
searches which will produce approximately 100 GB output (inclusive mzML
files)
usage:
./machine_offset_bruderer_sweep.py <folder_with_bruderer_data>
Sweeps over:
* machine_offset_in_ppm from -20 to +20 ppm offset
* precursor mass tolerance from 1 to 5 ppm
* fragment mass tolerance from -2.5 to 20 ppm
The search can be very time consuming (depending on your machine/cluster),
therefor the analyze step can be performed separately by calling analyze()
instead of search() when one has already performed the searches and wants
to analyze the results.
"""
file_2_target_offset = {}
for MQ_offset, file_name in MQ_OFFSET_TO_FILENAME:
file_2_target_offset[file_name] = {"offsets": []}
for n in range(-20, 20, 2):
file_2_target_offset[file_name]["offsets"].append(n)
engine_list = ["xtandem_sledgehammer"]
frag_ion_tolerance_list = [2.5, 5, 10, 20]
precursor_ion_tolerance_list = [1, 2, 3, 4, 5]
R = ursgal.UController(profile="QExactive+", params=GENERAL_PARAMS)
if os.path.exists(R.params["database"]) is False:
R.fetch_file(engine="get_http_files_1_0_0")
prefix_format_string = "_pit_{1}_fit_{2}"
for mzML_path in glob.glob(os.path.join(input_folder, "*.mzML")):
mzML_basename = os.path.basename(mzML_path)
if mzML_basename not in file_2_target_offset.keys():
continue
for ppm_offset in file_2_target_offset[mzML_basename]["offsets"]:
R.params["machine_offset_in_ppm"] = ppm_offset
R.params["prefix"] = "ppm_offset_{0}".format(int(ppm_offset))
mgf_file = R.convert(input_file=mzML_path, engine="mzml2mgf_1_0_0")
for engine in engine_list:
for precursor_ion_tolerane in precursor_ion_tolerance_list:
for frag_ion_tolerance in frag_ion_tolerance_list:
new_prefix = prefix_format_string.format(
precursor_ion_tolerane, frag_ion_tolerance
)
R.params[
"precursor_mass_tolerance_minus"
] = precursor_ion_tolerane
R.params[
"precursor_mass_tolerance_plus"
] = precursor_ion_tolerane
R.params["frag_mass_tolerance"] = frag_ion_tolerance
R.params["prefix"] = new_prefix
unified_search_result_file = R.search(
input_file=mzML_path,
engine=engine,
force=False,
)
return
def analyze(folder):
"""
Parses the result files form search and write a result .csv file which
contains the data to plot figure 2.
"""
R = ursgal.UController(profile="QExactive+", params=GENERAL_PARAMS)
csv_collector = {}
ve = "qvality_2_02"
sample_regex_pattern = "sample\d_R0\d"
sample_2_x_pos_and_mq_offset = {}
sample_offset_combos = []
all_tested_offsets = [str(n) for n in range(-20, 21, 2)]
for pos, (mq_ppm_off, mzML_file) in enumerate(MQ_OFFSET_TO_FILENAME):
_sample = re.search(sample_regex_pattern, mzML_file).group()
sample_2_x_pos_and_mq_offset[_sample] = (pos, mq_ppm_off)
for theo_offset in all_tested_offsets:
sample_offset_combos.append((_sample, theo_offset))
for csv_path in glob.glob(os.path.join("{0}".format(folder), "*", "*_unified.csv")):
dirname = os.path.dirname(csv_path)
sample = re.search(sample_regex_pattern, csv_path).group()
splitted_basename = os.path.basename(csv_path).split("_")
offset = splitted_basename[2]
precursor_ion_tolerance = splitted_basename[4]
frag_ion_tolerance = splitted_basename[6]
prefix = "_".join(splitted_basename[:7])
R.params["machine_offset_in_ppm"] = offset
R.params["precursor_mass_tolerance_minus"] = precursor_ion_tolerance
R.params["precursor_mass_tolerance_plus"] = precursor_ion_tolerance
R.params["frag_mass_tolerance"] = frag_ion_tolerance
R.params["prefix"] = prefix
validated_path = csv_path.replace(
"_unified.csv", "_{0}_validated.csv".format(ve)
)
if os.path.exists(validated_path):
csv_path = validated_path
else:
try:
csv_path = R.validate(input_file=csv_path, engine=ve)
except:
continue
pit_fit = (precursor_ion_tolerance, frag_ion_tolerance)
if pit_fit not in csv_collector.keys():
csv_collector[pit_fit] = ddict(set)
csv_key = (sample, offset)
print("Reading file: {0}".format(csv_path))
for line_dict in csv.DictReader(open(csv_path, "r")):
if line_dict["Is decoy"] == "true":
continue
if float(line_dict["PEP"]) <= 0.01:
csv_collector[pit_fit][csv_key].add(
"{0}{1}".format(line_dict["Sequence"], line_dict["Modifications"])
)
fieldnames = ["Sample", "pos", "MQ_offset", "tested_ppm_offset", "peptide_count"]
outfile_name_format_string = "bruderer_data_ppm_sweep_precursor_mass_tolerance_{0}_fragment_mass_tolerance_{1}.csv"
for pit_fit in csv_collector.keys():
with open(outfile_name_format_string.format(*pit_fit), "w") as io:
csv_writer = csv.DictWriter(io, fieldnames)
csv_writer.writeheader()
# write missing values
for sample_offset in sample_offset_combos:
sample, ppm_offset = sample_offset
if sample_offset not in csv_collector[pit_fit].keys():
dict_2_write = {
"Sample": sample,
"pos": sample_2_x_pos_and_mq_offset[sample][0],
"MQ_offset": "",
"tested_ppm_offset": ppm_offset,
"peptide_count": 0,
}
csv_writer.writerow(dict_2_write)
for (sample, ppm_offset), peptide_set in csv_collector[pit_fit].items():
dict_2_write = {
"Sample": sample,
"pos": sample_2_x_pos_and_mq_offset[sample][0],
"MQ_offset": sample_2_x_pos_and_mq_offset[sample][1] * -1,
"tested_ppm_offset": ppm_offset,
"peptide_count": len(peptide_set),
}
csv_writer.writerow(dict_2_write)
return
if __name__ == "__main__":
if len(sys.argv) < 2:
print(search.__doc__)
sys.exit(1)
search(sys.argv[1])
analyze(sys.argv[1])
Filter CSV Examples¶
Filter for modifications¶
-
filter_csv_for_mods_example.main()¶ Examples script for filtering unified results for modification containing entries
- usage:
- ./filter_csv_for_mods_example.py
Will produce a file with only entries which contain Carbamidomethyl as a modification.
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Examples script for filtering unified results for modification containing
entries
usage:
./filter_csv_for_mods_example.py
Will produce a file with only entries which contain Carbamidomethyl as a
modification.
"""
params = {
"csv_filter_rules": [
["Modifications", "contains", "Carbamidomethyl"],
],
"write_unfiltered_results": False,
}
csv_file_to_filter = os.path.join(
os.pardir,
"example_data",
"misc",
"filter_csv_for_mods_example_omssa_2_1_9_pmap_unified.csv",
)
uc = ursgal.UController(params=params)
filtered_csv = uc.execute_misc_engine(
input_file=csv_file_to_filter,
engine="filter_csv_1_0_0",
)
if __name__ == "__main__":
main()
Filter validated results¶
-
filter_csv_validation_example.main()¶ Examples script for filtering validated results for a PEP <= 0.01 and remove all decoys.
- usage:
- ./filter_csv_validation_example.py
Will produce a file with only target sequences with a posterior error probability of lower or equal to 1 percent
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import os
def main():
"""
Examples script for filtering validated results for a PEP <= 0.01 and
remove all decoys.
usage:
./filter_csv_validation_example.py
Will produce a file with only target sequences with a posterior error
probability of lower or equal to 1 percent
"""
params = {
"csv_filter_rules": [["PEP", "lte", 0.01], ["Is decoy", "equals", "false"]]
}
csv_file_to_filter = os.path.join(
os.pardir,
"example_data",
"misc",
"filter_csv_for_mods_example_omssa_2_1_9_pmap_unified_percolator_2_08_validated.csv",
)
uc = ursgal.UController(params=params)
filtered_csv = uc.execute_misc_engine(
input_file=csv_file_to_filter,
engine="filter_csv_1_0_0",
)
if __name__ == "__main__":
main()
SugarPy Example Scripts¶
SugarPy complete workflow¶
-
sugarpy_example_workflow.main(folder=None, target_decoy_database=None)¶ Complete example workflow for the identification of intact glycopeptides from MS runs employing IS-CID.
- usage:
- ./do_it_all_folder_wide.py <mzML_folder> <target_decoy_database>
#!/usr/bin/env python3.4
# encoding: utf-8
import ursgal
import sys
import glob
import os
import shutil
def main(folder=None, target_decoy_database=None):
'''
Complete example workflow for the identification of intact glycopeptides
from MS runs employing IS-CID.
usage:
./do_it_all_folder_wide.py <mzML_folder> <target_decoy_database>
'''
# define folder with mzML_files as sys.argv[1]
mzML_files = []
for mzml in glob.glob(os.path.join('{0}'.format(folder), '*.mzML')):
mzML_files.append(mzml)
mass_spectrometer = 'QExactive+'
# Define search engines for protein database search
search_engines = [
'xtandem_vengeance',
'msgfplus_v2019_07_03',
'msfragger_2_3',
]
# Define validation engine for protein database search
validation_engine = 'percolator_3_4_0'
# Modifications that should be included in the search.
# Glycan modifications will be added later.
all_mods = [
'C,fix,any,Carbamidomethyl',
'M,opt,any,Oxidation',
'*,opt,Prot-N-term,Acetyl',
]
# Initializing the Ursgal UController class with
# our specified modifications and mass spectrometer
params = {
'cpus':8,
'database' : target_decoy_database,
'modifications' : all_mods,
'csv_filter_rules' : [
['Is decoy', 'equals', 'false'],
['PEP', 'lte', 0.01],
['Conflicting uparam', 'contains_not', 'enzyme'],
],
'peptide_mapper_class_version': 'UPeptideMapper_v4',
'use_pyqms_for_mz_calculation': True,
'-xmx' : '16g',
'upper_mz_limit': 3000,
'lower_mz_limit': 500,
'precursor_mass_tolerance_plus': 5,
'precursor_mass_tolerance_minus': 5,
'frag_mass_tolerance': 20,
}
uc = ursgal.UController(
profile=mass_spectrometer,
params=params
)
# complete workflow
result_files_unfilered = []
glyco_result_files = []
pot_glyco_result_files = []
sugarpy_result_files = []
sugarpy_curated_files = []
for spec_file in mzML_files:
mgf_file = uc.convert(
input_file=spec_file,
engine='mzml2mgf_2_0_0'
)
results_one_file = []
for search_engine in search_engines:
# search MS2 specs for peptides with HexNAc and HexNAc(2) modification
for mod in [
'N,opt,any,HexNAc',
'N,opt,any,HexNAc(2)'
]:
uc.params['modifications'].append(mod)
uc.params['prefix'] = mod.split(',')[3]
search_result = uc.search_mgf(
input_file=mgf_file,
engine=search_engine
)
uc.params['prefix'] = ''
converted_result = uc.convert(
input_file=search_result,
guess_engine=True,
)
mapped_results = uc.execute_misc_engine(
input_file=converted_result,
engine='upeptide_mapper'
)
unified_search_results = uc.execute_misc_engine(
input_file=mapped_results,
engine='unify_csv',
)
validated_csv = uc.validate(
input_file=unified_search_results,
engine=validation_engine,
)
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_csv,
engine='filter_csv',
)
results_one_file.append(filtered_validated_results)
uc.params['modifications'].remove(mod)
uc.params['prefix'] = '{0}'.format(
os.path.basename(spec_file).strip('.mzML'))
all_results_one_file = uc.execute_misc_engine(
input_file=results_one_file,
engine='merge_csvs',
)
result_files_unfilered.append(all_results_one_file)
# filter for glycopeptides
uc.params.update({
'prefix': 'Glyco_',
'csv_filter_rules': [
['Modifications', 'contains', 'HexNAc'],
['Modifications', 'mod_at_glycosite', 'HexNAc'],
]
})
glyco_file = uc.execute_misc_engine(
input_file=all_results_one_file,
engine='filter_csv',
)
glyco_result_files.append(glyco_file)
# sugarpy_run to match glycopeptide fragments in MS1 scans.
# Modifiy those parameters according your MS specifications
uc.params.update({
'prefix': '',
'mzml_input_file': spec_file,
'min_number_of_spectra': 2,
'min_glycan_length': 3,
'max_glycan_length': 20,
'min_subtree_coverage': 0.65,
'min_sugarpy_score': 1,
'ms_level': 1,
'rt_border_tolerance': 1,
'precursor_max_charge': 5,
'use_median_accuracy': 'peptide',
'monosaccharide_compositions': {
"dHex": 'C6H10O4',
"Hex": 'C6H10O5',
"HexNAc": 'C8H13NO5',
"NeuAc": 'C11H17NO8',
# "dHexNAc": 'C8H13NO4',
# "HexA": 'C6H8O6',
# "Me2Hex": 'C8H14O5',
# "MeHex": 'C7H12O5',
# "Pent": 'C5H8O4',
# 'dHexN': 'C6H11O3N',
# 'HexN': 'C6H11O4N',
# 'MeHexA': 'C7H10O6',
},
'm_score_cutoff': 0.7,
'mz_score_percentile': 0.7,
'precursor_mass_tolerance_plus': 10,
'precursor_mass_tolerance_minus': 10,
'rel_intensity_range': 0.2,
'rt_pickle_name': os.path.join(
os.path.dirname(spec_file),
'_ursgal_lookup.pkl'
)
})
sugarpy_results = uc.execute_misc_engine(
input_file=glyco_file,
engine='sugarpy_run_1_0_0',
)
sugarpy_result_files.append(sugarpy_results)
# sugarpy_plot to plot the results from the sugarpy_run
# This will plot an elution profile for all identified glycopeptides
uc.params.update({
'sugarpy_results_pkl': sugarpy_results.replace('.csv', '.pkl'),
'sugarpy_results_csv': sugarpy_results,
'sugarpy_plot_types': [
'plot_glycan_elution_profile',
],
'plotly_layout': {
'font' : {
'family':'Arial',
# 'size': 20,
'color' : 'rgb(0, 0, 0)',
},
'autosize':True,
# 'width':1700,
# 'height':700,
# 'margin':{
# 'l':100,
# 'r':80,
# 't':100,
# 'b':60,
# },
'xaxis':{
'type':'linear',
'color':'rgb(0, 0, 0)',
'title_font':{
'family':'Arial',
'size':20,
'color':'rgb(0, 0, 0)',
},
'autorange':True,
# 'range':[0,1000.0],
'tickmode':'auto',
'showexponent':'all',
'exponentformat':'B',
'ticklen':5,
'tickwidth':1,
'tickcolor':'rgb(0, 0, 0)',
'ticks':'outside',
'showline':True,
'linecolor':'rgb(0, 0, 0)',
'mirror':False,
'showgrid':False,
},
'yaxis':{
'type':'linear',
'color':'rgb(0, 0, 0)',
'title':'Intensity',
'title_font':{
'family':'Arial',
'size':20,
'color':'rgb(0, 0, 0)',
},
'autorange':True,
# 'range':[0.0,100.0],
'showexponent':'all',
'exponentformat':'B',
'ticklen':5,
'tickwidth':1,
'tickcolor':'rgb(0, 0, 0)',
'ticks':'outside',
'showline':True,
'linecolor':'rgb(0, 0, 0)',
'mirror':False,
'showgrid':False,
},
'legend':{
'orientation':'h',
'traceorder':'normal',
},
'showlegend':True,
'paper_bgcolor':'rgba(0,0,0,0)',
'plot_bgcolor':'rgba(0,0,0,0)',
},
})
sugarpy_plots = uc.execute_misc_engine(
input_file=spec_file,
engine='sugarpy_plot_1_0_0',
# force = True,
)
# setting uparams back to original
uc.params.update({
'prefix': '',
'ms_level': 2,
'precursor_max_charge': 5,
'csv_filter_rules': [
['Is decoy', 'equals', 'false'],
['PEP', 'lte', 0.01],
],
'precursor_mass_tolerance_plus' : 5,
'precursor_mass_tolerance_minus': 5,
'rt_pickle_name': '_ursgal_lookup.pkl',
'frag_mass_tolerance': 20,
# 'rt_pickle_name': os.path.join(
# os.path.dirname(spec_file),
# '_ursgal_lookup.pkl'
# )
})
# combine results from multiple mzML files
results_all_files_unfiltered = uc.execute_misc_engine(
input_file=result_files_unfilered,
engine='merge_csvs',
)
glyco_results_all_files = uc.execute_misc_engine(
input_file=glyco_result_files,
engine='merge_csvs',
)
sugarpy_results_all_files = uc.execute_misc_engine(
input_file=sugarpy_result_files,
engine='merge_csvs',
)
# Extract the best matches from the raw SugarPy results.
# This is used for the automatic filtering described in the manuscript.
uc.params.update({
'prefix': '',
'ms_level': 1,
'min_number_of_spectra': 2,
'max_trees_per_spec': 1,
'sugarpy_results_pkl': None,
'sugarpy_results_csv': sugarpy_results_all_files,
'sugarpy_plot_types': [
'extract_best_matches',
],
'rt_pickle_name': 'create_new_lookup',
})
sugarpy_extracted = uc.execute_misc_engine(
# the specific spec file is not important here,
# this is just used to localize the scan2rt lookup
input_file=spec_file,
engine='sugarpy_plot_1_0_0',
force = False,
)
# Filter extracted results for glycopeptides that were identified
# in at least two replicates (i.e. two MS files)
uc.params['csv_filter_rules'] = [
['Num Raw Files', 'gte', 2],
]
extracted_filtered = uc.execute_misc_engine(
input_file=sugarpy_results_all_files.replace('.csv', '_extracted.csv'),
engine='filter_csv',
)
if __name__ == '__main__':
if len(sys.argv) < 3:
print(main.__doc__)
exit()
main(
folder=sys.argv[1],
target_decoy_database=sys.argv[2],
)
SugarPy plot annotated specs¶
-
sugarpy_plot_annotated_specs.main(spec_file=None, sugarpy_results=None, sugarpy_results_pkl=None)¶ This script can be used to plot any spectrum from SugarPy results. It woll plot all spectra listed in the input file (sugarpy_results), therefore it is recommended to provide a csv file with only a few, representative spectra for each glycopeptide. Besides the mzml file corresponding to the glycopeptide identifications, the sugarpy results csv file as well as the corresponding pkl file is required.
- Usage:
- ./sugarpy_plot_annotated_specs.py <mzml.idx.gz> <sugarpy_results.csv> <sugarpy_results_pkl>
#!/usr/bin/env python3.4
# encoding: utf-8
import ursgal
import sys
import glob
import os
import shutil
def main(spec_file=None, sugarpy_results=None, sugarpy_results_pkl=None):
'''
This script can be used to plot any spectrum from SugarPy results.
It woll plot all spectra listed in the input file (sugarpy_results),
therefore it is recommended to provide a csv file with only a few,
representative spectra for each glycopeptide.
Besides the mzml file corresponding to the glycopeptide identifications,
the sugarpy results csv file as well as the corresponding pkl file is required.
Usage:
./sugarpy_plot_annotated_specs.py <mzml.idx.gz> <sugarpy_results.csv> <sugarpy_results_pkl>
'''
# initialize the UController
uc = ursgal.UController()
# sugarpy_plot to plot he results from the sugarpy_run
uc.params.update({
'prefix': 'Annotated_specs',
'sugarpy_results_pkl': sugarpy_results_pkl,
'sugarpy_results_csv': sugarpy_results,
'sugarpy_plot_types': [
'plot_annotated_spectra',
],
'sugarpy_plot_peak_types': [
'matched',
'unmatched',
'labels',
],
# 'sugarpy_include_subtrees': 'individual_subtrees'
'rt_pickle_name': os.path.join(
os.path.dirname(spec_file),
'_ursgal_lookup.pkl'
),
'ms_level': 1,
'plotly_layout': {
'font' : {
'family':'Arial',
'size': 26,
'color' : 'rgb(0, 0, 0)',
},
# 'autosize':True,
'width':1750,
'height':820,
'margin':{
'l':120,
'r':50,
't':50,
'b':170,
},
'xaxis':{
'type':'linear',
'color':'rgb(0, 0, 0)',
'title': 'm/z',
'title_font':{
'family':'Arial',
'size':26,
'color':'rgb(0, 0, 0)',
},
'autorange':True,
# 'range':[1000,1550.0],
'tickmode':'auto',
'showticklabels':True,
'tickfont':{
'family':'Arial',
'size':22,
'color':'rgb(0, 0, 0)',
},
'showexponent':'all',
'exponentformat':'B',
'ticklen':5,
'tickwidth':1,
'tickcolor':'rgb(0, 0, 0)',
'ticks':'outside',
'showline':True,
'linecolor':'rgb(0, 0, 0)',
'showgrid':False,
'dtick':100,
},
'yaxis':{
'type':'linear',
'color':'rgb(0, 0, 0)',
'title':'Intensity',
'title_font':{
'family':'Arial',
'size':26,
'color':'rgb(0, 0, 0)',
},
'autorange':True,
# 'range':[0.0,25.0],
'showticklabels':True,
'tickfont':{
'family':'Arial',
'size':22,
'color':'rgb(0, 0, 0)',
},
'tickangle':0,
'showexponent':'all',
'exponentformat':'B',
'ticklen':5,
'tickwidth':1,
'tickcolor':'rgb(0, 0, 0)',
'ticks':'outside',
'showline':True,
'linecolor':'rgb(0, 0, 0)',
'showgrid':False,
'zeroline':False,
},
# 'legend':{
# 'font':{
# 'family':'Arial',
# 'size':10,
# 'color':'rgb(0, 0, 0)',
# },
# 'orientation':'v',
# 'traceorder':'normal',
# },
'showlegend':False,
'paper_bgcolor':'rgba(0,0,0,0)',
'plot_bgcolor':'rgba(0,0,0,0)',
},
})
sugarpy_plots = uc.execute_misc_engine(
input_file=spec_file,
engine='sugarpy_plot_1_0_0',
force = True,
)
if __name__ == '__main__':
main(
spec_file=sys.argv[1],
sugarpy_results=sys.argv[2],
)
Cascade Search¶
Cascade search example¶
-
cascade_search_example.search(validation_engine)¶ Executes a cascade search on four example files from the data from Barth et al.
- usage:
- ./cascade_search_example.py
Searches for peptides using a cascade search approach similar to Kertesz-Farkas et al. for which spectra were first searched for unmodified peptides, followed by consecutive searches for the following modifications: oxidation of M, deamidation of N/Q, methylation of E/K/R, N-terminal acetylation, phosphorylation of S/T. After each step, spectra with a PSM below 1 % PEP were removed.
-
cascade_search_example.analyze(collector)¶ Simle analysis script for the cascade search, counting the number of identified peptides (combination of peptide sequence and modifications) and PSMs (additionally include the spectrum ID)
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import csv
from collections import defaultdict as ddict
import os
import glob
import math
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
],
}
# We specify all search engines and validation engines that we want
# to use in a list (version numbers might differ on windows or mac):
search_engines = [
"omssa",
"xtandem_piledriver",
"msgfplus_v9979",
# 'myrimatch_2_1_138',
# 'msamanda_1_0_0_5243',
]
validation_engines = [
"percolator_2_08",
"qvality",
]
# The different levels with different modifications
# for the cascade are defined
cascade = {
"0": ["C,fix,any,Carbamidomethyl"],
"1": ["C,fix,any,Carbamidomethyl", "N,opt,any,Deamidated", "Q,opt,any,Deamidated"],
"2": ["C,fix,any,Carbamidomethyl", "*,opt,Prot-N-term,Acetyl"],
"3": ["C,fix,any,Carbamidomethyl", "M,opt,any,Oxidation"],
"4": [
"C,fix,any,Carbamidomethyl",
"E,opt,any,Methyl",
"K,opt,any,Methyl",
"R,opt,any,Methyl",
],
"5": ["C,fix,any,Carbamidomethyl", "S,opt,any,Phospho", "T,opt,any,Phospho"],
}
mass_spectrometer = "LTQ XL low res"
get_params = {
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
"JB_FASP_pH8_2-4_28122012.mzML",
"JB_FASP_pH8_3-1_28122012.mzML",
"JB_FASP_pH8_4-1_28122012.mzML",
],
"ftp_output_folder": os.path.join(os.pardir, "example_data", "cascade_search"),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
def get_files():
uc = ursgal.UController(params=get_params)
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
if os.path.exists(get_params["ftp_output_folder"]) is False:
os.makedirs(get_params["ftp_output_folder"])
uc.fetch_file(engine="get_ftp_files_1_0_0")
spec_files = []
for mzML_file in glob.glob(os.path.join(get_params["ftp_output_folder"], "*.mzML")):
spec_files.append(mzML_file)
return spec_files
def search(validation_engine):
"""
Executes a cascade search on four example files from the
data from Barth et al.
usage:
./cascade_search_example.py
Searches for peptides using a cascade search approach similar to Kertesz-Farkas et al.
for which spectra were first searched for unmodified peptides, followed by consecutive searches
for the following modifications:
oxidation of M,
deamidation of N/Q,
methylation of E/K/R,
N-terminal acetylation,
phosphorylation of S/T.
After each step, spectra with a PSM below 1 % PEP were removed.
"""
# Initializing the uPLANIT UController class with
# our specified modifications and mass spectrometer
uc = ursgal.UController(
profile=mass_spectrometer, params=params # 'LTQ XL low res' profile!
)
# complete workflow for every level of the cascade:
# every spectrum file is searched with every search engine,
# results are validated seperately,
# validated results are merged and filtered for targets and PEP <= 0.01.
def workflow(
spec_file,
prefix=None,
validation_engine=None,
filter_before_validation=False,
force=False,
):
validated_results = []
# Convert mzML to MGF outside the loop, so this step is not repeated in
# the loop
mgf_spec_file = uc.convert(input_file=spec_file, engine="mzml2mgf_1_0_0")
for search_engine in search_engines:
uc.params["prefix"] = prefix
unified_search_results = uc.search(
input_file=mgf_spec_file,
engine=search_engine,
force=force,
)
uc.params["prefix"] = ""
if filter_before_validation == True:
uc.params["csv_filter_rules"] = [
[
"Modifications",
"contains",
"{0}".format(cascade[level][1].split(",")[3]),
]
]
filtered_search_results = uc.execute_misc_engine(
input_file=unified_search_results, engine="filter_csv_1_0_0"
)
else:
filtered_search_results = unified_search_results
validated_search_results = uc.validate(
input_file=filtered_search_results,
engine=validation_engine,
force=force,
)
validated_results.append(validated_search_results)
validated_results_from_all_engines = uc.execute_misc_engine(
input_file=sorted(validated_results),
engine="merge_csvs_1_0_0",
force=force,
)
uc.params["csv_filter_rules"] = [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
]
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_results_from_all_engines, engine="filter_csv_1_0_0"
)
return filtered_validated_results
result_files = []
for spec_file in spec_files:
spectra_with_PSM = set()
for level in sorted(cascade.keys()):
uc.params["modifications"] = cascade[level]
if level == "0":
results = workflow(
spec_file,
validation_engine=validation_engine,
prefix="cascade-lvl-{0}".format(level),
)
else:
uc.params["scan_exclusion_list"] = list(spectra_with_PSM)
results = workflow(
spec_file,
validation_engine=validation_engine,
filter_before_validation=True,
force=True,
prefix="cascade-lvl-{0}".format(level),
)
result_files.append(results)
# spectrum IDs for PSMs are written into an exclusion list for the next level of the cascade search,
# these spectra will b excluded during mzml2mgf conversion
with open(results) as in_file:
csv_input = csv.DictReader(in_file)
for line_dict in csv_input:
spectra_with_PSM.add(line_dict["Spectrum ID"])
print(
"Number of spectra that will be removed for the next cacade level: {0}".format(
len(spectra_with_PSM)
)
)
results_all_files = uc.execute_misc_engine(
input_file=sorted(result_files),
engine="merge_csvs_1_0_0",
)
return results_all_files
def analyze(collector):
"""
Simle analysis script for the cascade search,
counting the number of identified peptides (combination of peptide sequence and modifications)
and PSMs (additionally include the spectrum ID)
"""
mod_list = ["Oxidation", "Deamidated", "Methyl", "Acetyl", "Phospho"]
fieldnames = (
["approach", "count_type", "validation_engine", "unmodified", "multimodified"]
+ mod_list
+ ["total"]
)
csv_writer = csv.DictWriter(open("cascade_results.csv", "w"), fieldnames)
csv_writer.writeheader()
uc = ursgal.UController()
uc.params["validation_score_field"] = "PEP"
uc.params["bigger_scores_better"] = False
# Count the number of identified peptides and PSMs for the different modifications
# Spectra with multiple PSMs are sanitized, i.e. only the PSM with best PEP score is counted
# and only if the best hit has a PEP that is at least two orders of
# magnitude smaller than the others
for validation_engine, result_file in collector.items():
counter_dict = {"psm": ddict(set), "pep": ddict(set)}
grouped_psms = uc._group_psms(
result_file, validation_score_field="PEP", bigger_scores_better=False
)
for spec_title, grouped_psm_list in grouped_psms.items():
best_score, best_line_dict = grouped_psm_list[0]
if len(grouped_psm_list) > 1:
second_best_score, second_best_line_dict = grouped_psm_list[1]
best_peptide_and_mod = (
best_line_dict["Sequence"] + best_line_dict["Modifications"]
)
second_best_peptide_and_mod = (
second_best_line_dict["Sequence"]
+ second_best_line_dict["Modifications"]
)
if best_peptide_and_mod == second_best_peptide_and_mod:
line_dict = best_line_dict
elif best_line_dict["Sequence"] == second_best_line_dict["Sequence"]:
if best_score == second_best_score:
line_dict = best_line_dict
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
line_dict = best_line_dict
count = 0
for mod in mod_list:
if mod in line_dict["Modifications"]:
count += 1
key_2_add = ""
if count == 0:
key_2_add = "unmodified"
elif count >= 2:
key_2_add = "multimodified"
elif count == 1:
for mod in mod_list:
if mod in line_dict["Modifications"]:
key_2_add = mod
break
# for peptide identification comparison
counter_dict["pep"][key_2_add].add(
line_dict["Sequence"] + line_dict["Modifications"]
)
# for PSM comparison
counter_dict["psm"][key_2_add].add(
line_dict["Spectrum Title"]
+ line_dict["Sequence"]
+ line_dict["Modifications"]
)
for counter_key, count_dict in counter_dict.items():
dict_2_write = {
"approach": "cascade",
"count_type": counter_key,
"validation_engine": validation_engine,
}
total_number = 0
for key, obj_set in count_dict.items():
dict_2_write[key] = len(obj_set)
total_number += len(obj_set)
dict_2_write["total"] = total_number
csv_writer.writerow(dict_2_write)
return
if __name__ == "__main__":
spec_files = get_files()
collector = {}
for validation_engine in validation_engines:
results_all_files = search(validation_engine)
print(
">>> ",
"final results for {0}".format(validation_engine),
" were written into:",
)
print(">>> ", results_all_files)
collector[validation_engine] = results_all_files
analyze(collector)
print(">>> ", "number of identified peptides and PSMs were written into:")
print(">>> ", "cascade_results.csv")
Ungrouped search for comparison¶
-
ungrouped_search_example.search(validation_engine)¶ Executes an ungrouped search on four example files from the data from Barth et al.
- usage:
- ./ungrouped_search_example.py
Searches for peptides including the following potential modifications: oxidation of M, deamidation of N/Q, methylation of E/K/R, N-terminal acetylation, phosphorylation of S/T.
All modifications are validated together with unmodified peptides.
-
ungrouped_search_example.analyze(collector)¶ Simple analysis script for the ungrouped search, counting the number of identified peptides (combination of peptide sequence and modifications) and PSMs (additionally include the spectrum ID)
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import csv
from collections import defaultdict as ddict
import os
import glob
import math
import sys
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
],
# Modifications that should be included in the search
"modifications": [
"C,fix,any,Carbamidomethyl",
"M,opt,any,Oxidation",
"N,opt,any,Deamidated",
"Q,opt,any,Deamidated",
"E,opt,any,Methyl",
"K,opt,any,Methyl",
"R,opt,any,Methyl",
"*,opt,Prot-N-term,Acetyl",
"S,opt,any,Phospho",
"T,opt,any,Phospho",
],
}
# We specify all search engines and validation engines that we want
# to use in a list (version numbers might differ on windows or mac):
search_engines = [
"omssa",
"xtandem_piledriver",
"msgfplus_v9979",
# 'myrimatch_2_1_138',
"msamanda_1_0_0_5243",
]
validation_engines = [
"percolator_2_08",
"qvality",
]
mass_spectrometer = "LTQ XL low res"
get_params = {
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
"JB_FASP_pH8_2-4_28122012.mzML",
"JB_FASP_pH8_3-1_28122012.mzML",
"JB_FASP_pH8_4-1_28122012.mzML",
],
"ftp_output_folder": os.path.join(os.pardir, "example_data", "ungrouped_search"),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
def get_files():
uc = ursgal.UController(params=get_params)
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
if os.path.exists(get_params["ftp_output_folder"]) is False:
os.makedirs(get_params["ftp_output_folder"])
uc.fetch_file(engine="get_ftp_files_1_0_0")
spec_files = []
for mzML_file in glob.glob(os.path.join(get_params["ftp_output_folder"], "*.mzML")):
spec_files.append(mzML_file)
return spec_files
def search(validation_engine):
"""
Executes an ungrouped search on four example files from the
data from Barth et al.
usage:
./ungrouped_search_example.py
Searches for peptides including the following potential modifications:
oxidation of M,
deamidation of N/Q,
methylation of E/K/R,
N-terminal acetylation,
phosphorylation of S/T.
All modifications are validated together with unmodified peptides.
"""
uc = ursgal.UController(
profile=mass_spectrometer, params=params # 'LTQ XL low res' profile!
)
# complete workflow:
# every spectrum file is searched with every search engine,
# results are validated seperately,
# validated results are merged and filtered for targets and PEP <= 0.01.
# In the end, all filtered results from all spectrum files are merged
# for validation_engine in validation_engines:
result_files = []
for spec_file in spec_files:
validated_results = []
for search_engine in search_engines:
unified_search_results = uc.search(
input_file=spec_file,
engine=search_engine,
)
validated_search_results = uc.validate(
input_file=unified_search_results,
engine=validation_engine,
)
validated_results.append(validated_search_results)
validated_results_from_all_engines = uc.execute_misc_engine(
input_file=sorted(validated_results),
engine="merge_csvs",
)
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_results_from_all_engines, engine="filter_csv"
)
result_files.append(filtered_validated_results)
results_all_files = uc.execute_misc_engine(
input_file=sorted(result_files),
engine="merge_csvs",
)
return results_all_files
def analyze(collector):
"""
Simple analysis script for the ungrouped search,
counting the number of identified peptides (combination of peptide sequence and modifications)
and PSMs (additionally include the spectrum ID)
"""
mod_list = ["Oxidation", "Deamidated", "Methyl", "Acetyl", "Phospho"]
fieldnames = (
["approach", "count_type", "validation_engine", "unmodified", "multimodified"]
+ mod_list
+ ["total"]
)
csv_writer = csv.DictWriter(open("ungrouped_results.csv", "w"), fieldnames)
csv_writer.writeheader()
uc = ursgal.UController()
uc.params["validation_score_field"] = "PEP"
uc.params["bigger_scores_better"] = False
# Count the number of identified peptides and PSMs for the different modifications
# Spectra with multiple PSMs are sanitized, i.e. only the PSM with best PEP score is counted
# and only if the best hit has a PEP that is at least two orders of
# magnitude smaller than the others
for validation_engine, result_file in collector.items():
counter_dict = {"psm": ddict(set), "pep": ddict(set)}
grouped_psms = uc._group_psms(
result_file, validation_score_field="PEP", bigger_scores_better=False
)
for spec_title, grouped_psm_list in grouped_psms.items():
best_score, best_line_dict = grouped_psm_list[0]
if len(grouped_psm_list) > 1:
second_best_score, second_best_line_dict = grouped_psm_list[1]
best_peptide_and_mod = (
best_line_dict["Sequence"] + best_line_dict["Modifications"]
)
second_best_peptide_and_mod = (
second_best_line_dict["Sequence"]
+ second_best_line_dict["Modifications"]
)
if best_peptide_and_mod == second_best_peptide_and_mod:
line_dict = best_line_dict
elif best_line_dict["Sequence"] == second_best_line_dict["Sequence"]:
if best_score == second_best_score:
line_dict = best_line_dict
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
line_dict = best_line_dict
count = 0
for mod in mod_list:
if mod in line_dict["Modifications"]:
count += 1
key_2_add = ""
if count == 0:
key_2_add = "unmodified"
elif count >= 2:
key_2_add = "multimodified"
elif count == 1:
for mod in mod_list:
if mod in line_dict["Modifications"]:
key_2_add = mod
break
# for peptide identification comparison
counter_dict["pep"][key_2_add].add(
line_dict["Sequence"] + line_dict["Modifications"]
)
# for PSM comparison
counter_dict["psm"][key_2_add].add(
line_dict["Spectrum Title"]
+ line_dict["Sequence"]
+ line_dict["Modifications"]
)
for counter_key, count_dict in counter_dict.items():
dict_2_write = {
"approach": "ungrouped",
"count_type": counter_key,
"validation_engine": validation_engine,
}
total_number = 0
for key, obj_set in count_dict.items():
dict_2_write[key] = len(obj_set)
total_number += len(obj_set)
dict_2_write["total"] = total_number
csv_writer.writerow(dict_2_write)
return
if __name__ == "__main__":
spec_files = get_files()
collector = {}
for validation_engine in validation_engines:
results_all_files = search(validation_engine)
print(
">>> ",
"final results for {0}".format(validation_engine),
" were written into:",
)
print(">>> ", results_all_files)
collector[validation_engine] = results_all_files
analyze(collector)
print(">>> ", "number of identified peptides and PSMs were written into:")
print(">>> ", "ungrouped_results.csv")
Grouped search for comparison¶
-
grouped_search_example.search(validation_engine)¶ Executes a grouped search on four example files from the data from Barth et al.
- usage:
- ./grouped_search_example.py
Searches for peptides including the following potential modifications: oxidation of M, deamidation of N/Q, methylation of E/K/R, N-terminal acetylation, phosphorylation of S/T.
After the search, each type of modification is validated seperately.
-
grouped_search_example.analyze(collector)¶ Simle analysis script for the grouped search, counting the number of identified peptides (combination of peptide sequence and modifications) and PSMs (additionally include the spectrum ID)
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import csv
from collections import defaultdict as ddict
import os
import glob
import math
from collections import defaultdict as ddict
params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"csv_filter_rules": [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
],
# Modifications that should be included in the search
"modifications": [
"C,fix,any,Carbamidomethyl",
"M,opt,any,Oxidation",
"N,opt,any,Deamidated",
"Q,opt,any,Deamidated",
"E,opt,any,Methyl",
"K,opt,any,Methyl",
"R,opt,any,Methyl",
"*,opt,Prot-N-term,Acetyl",
"S,opt,any,Phospho",
"T,opt,any,Phospho",
],
}
# We specify all search engines and validation engines that we want
# to use in a list (version numbers might differ on windows or mac):
search_engines = [
"omssa",
"xtandem_piledriver",
"msgfplus_v9979",
# 'myrimatch_2_1_138',
"msamanda_1_0_0_5243",
]
validation_engines = [
"percolator_2_08",
"qvality",
]
# Groups that are evaluated seperately
groups = {
"0": "",
"1": "Oxidation",
"2": "Deamidated",
"3": "Methyl",
"4": "Acetyl",
"5": "Phospho",
}
mass_spectrometer = "LTQ XL low res"
get_params = {
"ftp_url": "ftp.peptideatlas.org",
"ftp_login": "PASS00269",
"ftp_password": "FI4645a",
"ftp_include_ext": [
"JB_FASP_pH8_2-3_28122012.mzML",
"JB_FASP_pH8_2-4_28122012.mzML",
"JB_FASP_pH8_3-1_28122012.mzML",
"JB_FASP_pH8_4-1_28122012.mzML",
],
"ftp_output_folder": os.path.join(os.pardir, "example_data", "grouped_search"),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(os.pardir, "example_data"),
}
def get_files():
uc = ursgal.UController(params=get_params)
if os.path.exists(params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
if os.path.exists(get_params["ftp_output_folder"]) is False:
os.makedirs(get_params["ftp_output_folder"])
uc.fetch_file(engine="get_ftp_files_1_0_0")
spec_files = []
for mzML_file in glob.glob(os.path.join(get_params["ftp_output_folder"], "*.mzML")):
spec_files.append(mzML_file)
return spec_files
def search(validation_engine):
"""
Executes a grouped search on four example files from the
data from Barth et al.
usage:
./grouped_search_example.py
Searches for peptides including the following potential modifications:
oxidation of M,
deamidation of N/Q,
methylation of E/K/R,
N-terminal acetylation,
phosphorylation of S/T.
After the search, each type of modification is validated seperately.
"""
# Initializing the ursgal UController class with
# our specified modifications and mass spectrometer
uc = ursgal.UController(
profile=mass_spectrometer, params=params # 'LTQ XL low res' profile!
)
# complete workflow:
# every spectrum file is searched with every search engine,
# results are seperated into groups and validated seperately,
# validated results are merged and filtered for targets and PEP <= 0.01.
# In the end, all filtered results from all spectrum files are merged
# for validation_engine in validation_engines:
result_files = []
for n, spec_file in enumerate(spec_files):
validated_results = []
for search_engine in search_engines:
unified_search_results = uc.search(
input_file=spec_file,
engine=search_engine,
)
# Calculate PEP for every group seperately, therefore need to split
# the csv first
group_list = sorted(groups.keys())
for p, group in enumerate(group_list):
if group == "0":
uc.params["csv_filter_rules"] = [
["Modifications", "contains_not", "{0}".format(groups["1"])],
["Modifications", "contains_not", "{0}".format(groups["2"])],
["Modifications", "contains_not", "{0}".format(groups["3"])],
["Modifications", "contains_not", "{0}".format(groups["4"])],
["Modifications", "contains_not", "{0}".format(groups["5"])],
]
else:
uc.params["csv_filter_rules"] = [
["Modifications", "contains", "{0}".format(groups[group])]
]
for other_group in group_list:
if other_group == "0" or other_group == group:
continue
uc.params["csv_filter_rules"].append(
[
"Modifications",
"contains_not",
"{0}".format(groups[other_group]),
],
)
uc.params["prefix"] = "grouped-{0}".format(group)
filtered_results = uc.execute_misc_engine(
input_file=unified_search_results, engine="filter_csv"
)
uc.params["prefix"] = ""
validated_search_results = uc.validate(
input_file=filtered_results,
engine=validation_engine,
)
validated_results.append(validated_search_results)
uc.params["prefix"] = "file{0}".format(n)
validated_results_from_all_engines = uc.execute_misc_engine(
input_file=sorted(validated_results),
engine="merge_csvs",
)
uc.params["prefix"] = ""
uc.params["csv_filter_rules"] = [
["Is decoy", "equals", "false"],
["PEP", "lte", 0.01],
]
filtered_validated_results = uc.execute_misc_engine(
input_file=validated_results_from_all_engines, engine="filter_csv"
)
result_files.append(filtered_validated_results)
results_all_files = uc.execute_misc_engine(
input_files=sorted(result_files),
engine="merge_csvs",
)
return results_all_files
def analyze(collector):
"""
Simle analysis script for the grouped search,
counting the number of identified peptides (combination of peptide sequence and modifications)
and PSMs (additionally include the spectrum ID)
"""
mod_list = ["Oxidation", "Deamidated", "Methyl", "Acetyl", "Phospho"]
fieldnames = (
["approach", "count_type", "validation_engine", "unmodified", "multimodified"]
+ mod_list
+ ["total"]
)
csv_writer = csv.DictWriter(open("grouped_results.csv", "w"), fieldnames)
csv_writer.writeheader()
uc = ursgal.UController()
uc.params["validation_score_field"] = "PEP"
uc.params["bigger_scores_better"] = False
# Count the number of identified peptides and PSMs for the different modifications
# Spectra with multiple PSMs are sanitized, i.e. only the PSM with best PEP score is counted
# and only if the best hit has a PEP that is at least two orders of
# magnitude smaller than the others
for validation_engine, result_file in collector.items():
counter_dict = {"psm": ddict(set), "pep": ddict(set)}
grouped_psms = uc._group_psms(
result_file, validation_score_field="PEP", bigger_scores_better=False
)
for spec_title, grouped_psm_list in grouped_psms.items():
best_score, best_line_dict = grouped_psm_list[0]
if len(grouped_psm_list) > 1:
second_best_score, second_best_line_dict = grouped_psm_list[1]
best_peptide_and_mod = (
best_line_dict["Sequence"] + best_line_dict["Modifications"]
)
second_best_peptide_and_mod = (
second_best_line_dict["Sequence"]
+ second_best_line_dict["Modifications"]
)
if best_peptide_and_mod == second_best_peptide_and_mod:
line_dict = best_line_dict
elif best_line_dict["Sequence"] == second_best_line_dict["Sequence"]:
if best_score == second_best_score:
line_dict = best_line_dict
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
if (-1 * math.log10(best_score)) - (
-1 * math.log10(second_best_score)
) >= 2:
line_dict = best_line_dict
else:
continue
else:
line_dict = best_line_dict
count = 0
for mod in mod_list:
if mod in line_dict["Modifications"]:
count += 1
key_2_add = ""
if count == 0:
key_2_add = "unmodified"
elif count >= 2:
key_2_add = "multimodified"
elif count == 1:
for mod in mod_list:
if mod in line_dict["Modifications"]:
key_2_add = mod
break
# for peptide identification comparison
counter_dict["pep"][key_2_add].add(
line_dict["Sequence"] + line_dict["Modifications"]
)
# for PSM comparison
counter_dict["psm"][key_2_add].add(
line_dict["Spectrum Title"]
+ line_dict["Sequence"]
+ line_dict["Modifications"]
)
for counter_key, count_dict in counter_dict.items():
dict_2_write = {
"approach": "grouped",
"count_type": counter_key,
"validation_engine": validation_engine,
}
total_number = 0
for key, obj_set in count_dict.items():
dict_2_write[key] = len(obj_set)
total_number += len(obj_set)
dict_2_write["total"] = total_number
csv_writer.writerow(dict_2_write)
return
if __name__ == "__main__":
spec_files = get_files()
collector = {}
for validation_engine in validation_engines:
results_all_files = search(validation_engine)
print(
">>> ",
"final results for {0}".format(validation_engine),
" were written into:",
)
print(">>> ", results_all_files)
collector[validation_engine] = results_all_files
analyze(collector)
print(">>> ", "number of identified peptides and PSMs were written into:")
print(">>> ", "grouped_results.csv")
Example results for cascade search¶
ROS dataset results from a subsset of data from Barth et al. 2014.
| approach | count_type | validation_engine | unmodified | multimodified | Oxidation | Deamidated | Methyl | Acetyl | Phospho | total |
|---|---|---|---|---|---|---|---|---|---|---|
| ungrouped | psm | qvality | 6905 | 24 | 270 | 157 | 42 | 23 | 6 | 7427 |
| grouped | psm | qvality | 8009 | 267 | 128 | 31 | 27 | 3 | 8465 | |
| cascade | psm | qvality | 8009 | 316 | 131 | 46 | 30 | 1 | 7788 | |
| ungrouped | upep | qvality | 1409 | 14 | 94 | 48 | 20 | 11 | 3 | 1599 |
| grouped | upep | qvality | 1575 | 93 | 42 | 9 | 17 | 1 | 1737 | |
| cascade | upep | qvality | 1552 | 102 | 44 | 13 | 13 | 1 | 1725 |
Human BR dataset results.
| approach | count_type | validation_engine | unmodified | multimodified | Oxidation | Deamidated | Methyl | Acetyl | Phospho | total |
|---|---|---|---|---|---|---|---|---|---|---|
| ungrouped | psm | qvality | 18813 | 51 | 332 | 336 | 102 | 292 | 122 | 20048 |
| grouped | psm | qvality | 21370 | 383 | 171 | 100 | 435 | 142 | 22601 | |
| cascade | psm | qvality | 19065 | 424 | 251 | 111 | 417 | 38 | 20406 | |
| ungrouped | upep | qvality | 6707 | 39 | 143 | 173 | 27 | 123 | 58 | 7270 |
| grouped | upep | qvality | 7525 | 161 | 104 | 31 | 191 | 58 | 8070 | |
| cascade | upep | qvality | 7784 | 172 | 152 | 43 | 171 | 55 | 8377 |
Upeptide Mapper Example Scripts¶
Upeptide mapper v3 and 4 complete C. reinhardtii proteome match¶
-
complete_chlamydomonas_proteome_match.main(class_version)¶ Example script to demonstrate speed and memory efficiency of the new upeptide_mapper.
All tryptic peptides (n=1,094,395, 6 < len(peptide) < 40 ) are mapped to the Chlamydomonas reinhardtii (38876 entries) target-decoy database.
- usage:
- ./complete_chlamydomonas_proteome_match.py <class_version>
- Class versions
- UPeptideMapper_v2
- UPeptideMapper_v3
- UPeptideMapper_v4
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import os.path
import sys
import time
def main(class_version):
"""
Example script to demonstrate speed and memory efficiency of the new
upeptide_mapper.
All tryptic peptides (n=1,094,395, 6 < len(peptide) < 40 ) are mapped to the
Chlamydomonas reinhardtii (38876 entries) target-decoy database.
usage:
./complete_chlamydomonas_proteome_match.py <class_version>
Class versions
* UPeptideMapper_v2
* UPeptideMapper_v3
* UPeptideMapper_v4
"""
input_params = {
"database": os.path.join(
os.pardir,
"example_data",
"Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
),
"http_url": "https://www.sas.upenn.edu/~sschulze/Creinhardtii_281_v5_5_CP_MT_with_contaminants_target_decoy.fasta",
"http_output_folder": os.path.join(
os.pardir,
"example_data",
),
}
uc = ursgal.UController(params=input_params)
if os.path.exists(input_params["database"]) is False:
uc.fetch_file(engine="get_http_files_1_0_0")
print("Parsing fasta and digesting sequences")
peptides = set()
digest_start = time.time()
for fastaID, sequence in ursgal.ucore.parse_fasta(
open(input_params["database"], "r")
):
tryptic_peptides = ursgal.ucore.digest(
sequence, ("KR", "C"), no_missed_cleavages=True
)
for p in tryptic_peptides:
if 6 <= len(p) <= 40:
peptides.add(p)
print(
"Parsing fasta and digesting sequences took {0:1.2f} seconds".format(
time.time() - digest_start
)
)
if sys.platform == "win32":
print(
"[ WARNING ] pyahocorasick can not be installed via pip on Windwows at the moment\n"
"[ WARNING ] Falling back to UpeptideMapper_v2"
)
class_version = "UPeptideMapper_v2"
upapa_class = uc.unodes["upeptide_mapper_1_0_0"][
"class"
].import_engine_as_python_function(class_version)
print("Buffering fasta and mapping {0} peptides".format(len(peptides)))
map_start = time.time()
if class_version == "UPeptideMapper_v2":
peptide_mapper = upapa_class(word_len=6)
fasta_lookup_name = peptide_mapper.build_lookup_from_file(
input_params["database"],
force=False,
)
args = [list(peptides), fasta_lookup_name]
elif class_version == "UPeptideMapper_v3":
peptide_mapper = upapa_class(input_params["database"])
fasta_lookup_name = peptide_mapper.fasta_name
args = [list(peptides), fasta_lookup_name]
elif class_version == "UPeptideMapper_v4":
peptide_mapper = upapa_class(input_params["database"])
args = [list(peptides)]
p2p_mappings = peptide_mapper.map_peptides(*args)
print(
"Buffering fasta and mapping {0} peptides took {1:1.2f} seconds".format(
len(peptides), time.time() - map_start
)
)
if len(p2p_mappings.keys()) == len(peptides):
print("All peptides have been mapped!")
else:
print("WARNING: Not all peptide have been mapped")
if __name__ == "__main__":
if len(sys.argv) < 2:
print(main.__doc__)
sys.exit(1)
main(sys.argv[1])
Upeptide mapper v3 and 4 complete proteome match¶
-
complete_proteome_match.main(fasta_database, class_version)¶ Example script to demonstrate speed and memory efficiency of the new upeptide_mapper.
Specify fasta_database and class_version as input.
- usage:
- ./complete_proteome_match.py <fasta_database> <class_version>
- Class versions
- UPeptideMapper_v2
- UPeptideMapper_v3
- UPeptideMapper_v4
#!/usr/bin/env python3
# encoding: utf-8
import ursgal
import glob
import os.path
import sys
import time
def main(fasta_database, class_version):
"""
Example script to demonstrate speed and memory efficiency of the new
upeptide_mapper.
Specify fasta_database and class_version as input.
usage:
./complete_proteome_match.py <fasta_database> <class_version>
Class versions
* UPeptideMapper_v2
* UPeptideMapper_v3
* UPeptideMapper_v4
"""
input_params = {
"database": sys.argv[1],
}
uc = ursgal.UController(params=input_params)
print("Parsing fasta and digesting sequences")
peptides = set()
digest_start = time.time()
for fastaID, sequence in ursgal.ucore.parse_fasta(
open(input_params["database"], "r")
):
tryptic_peptides = ursgal.ucore.digest(
sequence,
("KR", "C"),
# no_missed_cleavages = True
)
for p in tryptic_peptides:
if 6 <= len(p) <= 40:
peptides.add(p)
print(
"Parsing fasta and digesting sequences took {0:1.2f} seconds".format(
time.time() - digest_start
)
)
if sys.platform == "win32":
print(
"[ WARNING ] pyahocorasick can not be installed via pip on Windwows at the moment\n"
"[ WARNING ] Falling back to UpeptideMapper_v2"
)
class_version = "UPeptideMapper_v2"
upapa_class = uc.unodes["upeptide_mapper_1_0_0"][
"class"
].import_engine_as_python_function(class_version)
print("Buffering fasta and mapping {0} peptides".format(len(peptides)))
map_start = time.time()
if class_version == "UPeptideMapper_v2":
peptide_mapper = upapa_class(word_len=6)
fasta_lookup_name = peptide_mapper.build_lookup_from_file(
input_params["database"],
force=False,
)
args = [list(peptides), fasta_lookup_name]
elif class_version == "UPeptideMapper_v3":
peptide_mapper = upapa_class(input_params["database"])
fasta_lookup_name = peptide_mapper.fasta_name
args = [list(peptides), fasta_lookup_name]
elif class_version == "UPeptideMapper_v4":
peptide_mapper = upapa_class(input_params["database"])
args = [list(peptides)]
p2p_mappings = peptide_mapper.map_peptides(*args)
print(
"Buffering fasta and mapping {0} peptides took {1:1.2f} seconds".format(
len(peptides), time.time() - map_start
)
)
if len(p2p_mappings.keys()) == len(peptides):
print("All peptides have been mapped!")
else:
print("WARNING: Not all peptide have been mapped")
if __name__ == "__main__":
if len(sys.argv) < 3:
print(main.__doc__)
sys.exit(1)
main(sys.argv[1], sys.argv[2])